
Structure. Despite tremendous advances in understanding its genetic underpinnings in the last few years, electrical status epilepticus during slow-wave sleep (ESES) is a poorly understood neurodevelopmental disorder and to a certain extent the prototype of an epileptic encephalopathy. Slow-wave sleep in affected children is entirely replaced by epileptiform activity, leading to significant neurocognitive impairment with an emphasis on speech impairment. In a recent publication in Annals of Neurology, alterations in CNKSR2 are identified in families with a more severe course of ESES, highlighting the postsynapse as a possible player in ESES pathogenesis.
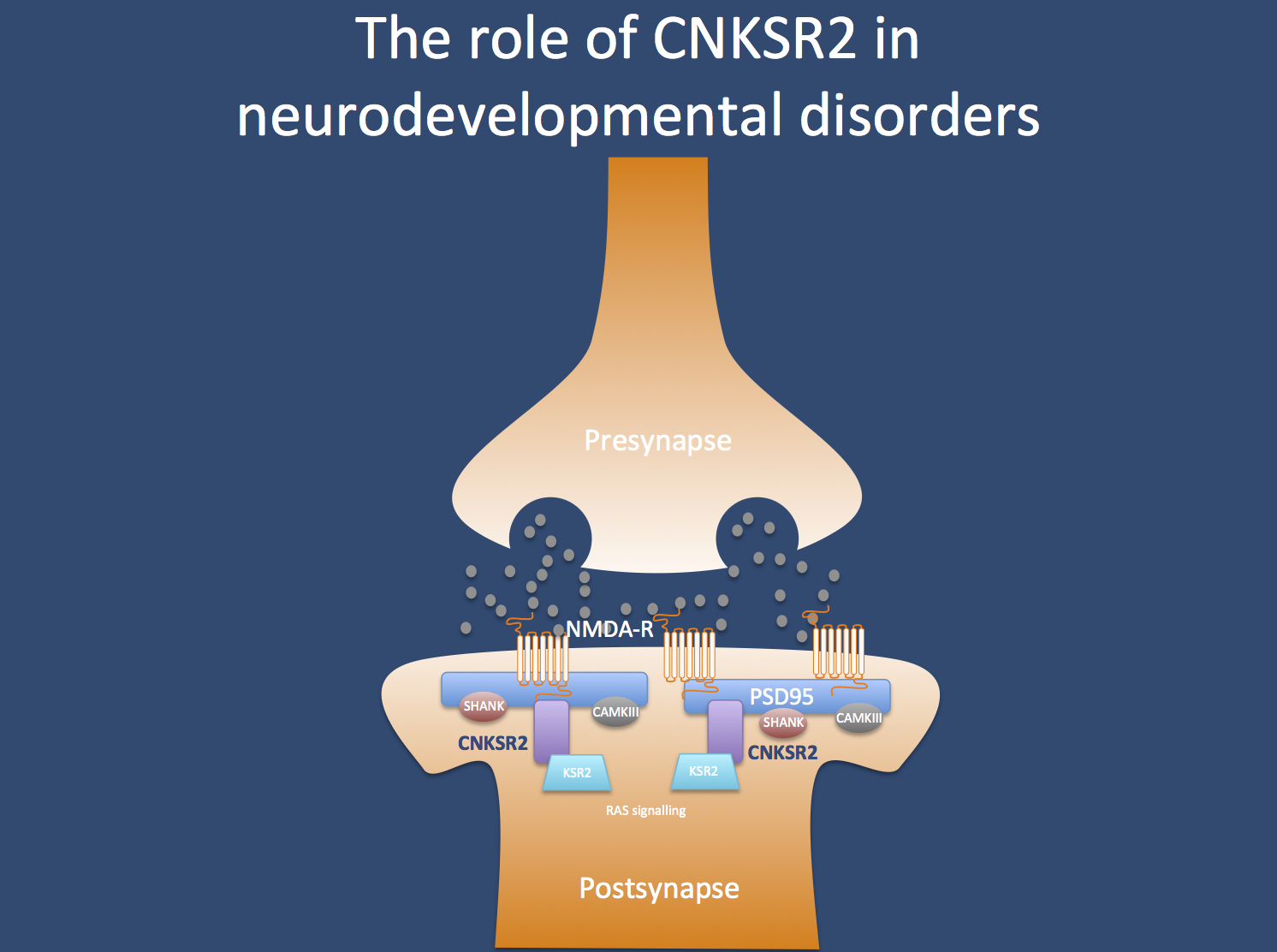
The CNKSR2 protein in the postsynapse.
ESES. The epileptic encephalopathy of ESES and its association with human speech is best understood by considering its part of a broader spectrum, the epilepsy-aphasia spectrum (EAS). Benign Rolandic Epilepsy can be considered the mildest form of EAS and is characterized by epileptiform discharges in the rolandic area, which is relevant in speech and motor activity associated with speech. If the epileptic discharges are more frequent, patients may have more severe epilepsy syndromes such as Pseudo-Lennox-Syndrome or Landau-Kleffner Syndrome. ESES represents the EEG finding when the epileptiform discharges in sleep are so prominent that they basically interfere with normal sleep architecture. Patients with ESES do not necessarily have seizures, but they usually have impairment of speech. In the most extreme scenarios, previously healthy children suddenly stop speaking (they become aphasic), and aggressive treatment of the ESES pattern on EEG with antiepileptic drugs may help them regain their ability to speak.
Genetics of ESES. The EEG feature of ESES and the associated speech impairment can be due to a variety of etiologies, ranging from brain lesions to genetic causes. As one of the most common genetic causes, GRIN2A mutations have been identified. In the publication by Vaags and collaborators, eight deletions and a mutation in CNKSR2 have been identified in four families with a particular ESES-related phenotype. CNKSR2 codes for the postsynaptic protein connector enhancer of KSR2, a structural protein of the postsynapse.
CNKSR2. The protein encoded by CNKSR2 does not have a very intuitive name: it codes for the connector enhancer of kinase suppressor of ras 2, also known as CNK homolog protein 2 (CNK2) or maguin (membrane-associated guanylate kinase-interacting protein). Basically, the CNSKR2 protein is located in the postsynapse, where it probably is the link between the postsynpatic structural proteins such as PSD95 and signaling cascades that are important for the growth of neuronal dendrites. Therefore, it is easily conceivable that alterations in the anchoring apparatus of the postsynapse may result in neurodevelopmental disorders.
The CNKSR2 phenotype. When looking at the phenotypes of the patients at more detail, a peculiar pattern emerges. Patients with CNKSR2 encephalopathy have some mild, yet non-specific developmental delay until the epilepsy starts at the age of 2 years. At this point, patients have ESES and regression of speech. However, in contrast to other patients with ESES, the course of the regression leads to overall stagnation of development and complete loss of speech that is not regained afterwards. Epilepsy is usually mild in patients with CNKSR2 encephalopathy, demonstrating again that ESES is not necessarily connected with a severe epilepsy phenotype. Patients usually have intellectual disability as a consequence of this and some patients have microcephaly. This protracted and stagnating course is usually more severe than seen in patients with ESES and complete loss of speech despite resolution of ESES in combination with intellectual disability may suggest a CNKSR2 phenotype.
What you need to know about CNKSR2. The CNKSR2 gene is a novel cause for a severe form of ESES, and further delineation of the phenotype may help decide what aspects of the phenotype are specific. It will also be of interest whether CNKSR2 alterations are found in patients with ESES with a less severe phenotype. Either way, the publication by Vaags and collaborators highlights the importance of the postsynapse in diseases of the epilepsy aphasia spectrum.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
