
SCN8A. In 2015, SCN8A has emerged as an important gene in epileptic encephalopathy. SCN8A encodes the voltage-gated sodium channel alpha subunit Nav1.6, and was first implicated in epileptic encephalopathy in 2012. Since then, approximately 100 cases of early-infantile epileptic encephalopathy caused by mutations of SCN8A have been identified, and the disorder has been designated EIEE13. Here is what you need to know about SCN8A in 2015.
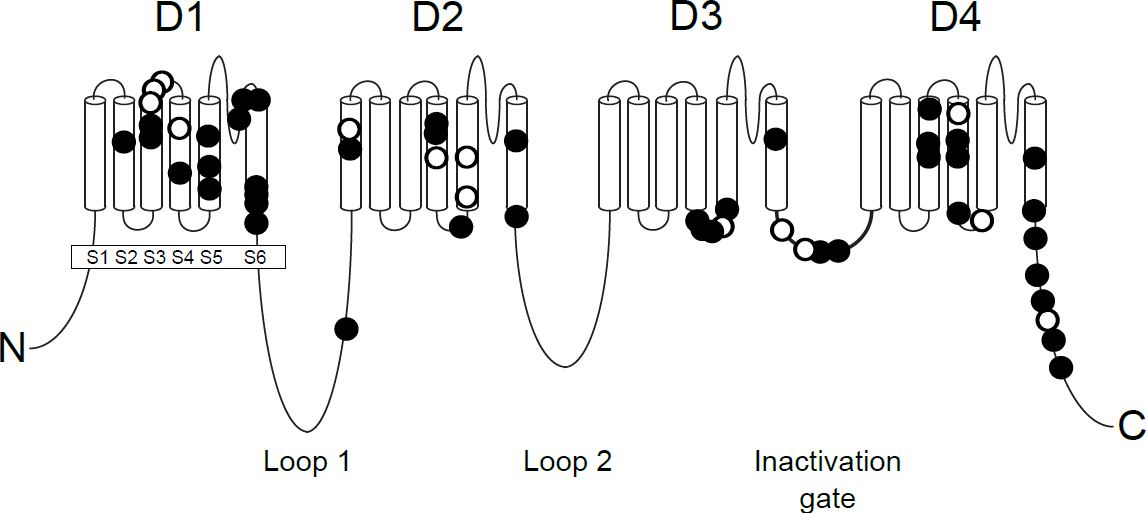
SCN8A encodes the voltage-gated sodium channel alpha subunit Nav1.6. Adapted from Wagnon and Meisler, Front Neurol 2015.
Phenotypes
EIEE13. Mutations in SCN8A cause an early-infantile epileptic encephalopathy designated EIEE13. EIEE13 is characterized by seizure onset between 0-18 months of age (mean of 5 months), mild to severe intellectual disability, and developmental delay with or without regression. Seizures are frequently polymorphic. Generalized tonic-clonic seizures occur in many patients, and tonic, atonic, myoclonic, focal and absence seizures are not uncommon.
In contrast with Dravet syndrome, febrile seizures are rarely observed in EIEE13. Seizures are generally refractory to treatment, but sodium channel blockers including carbamazepine, oxcarbazepine, and phenytoin have been used successfully in several patients. EEG findings often comprise moderate to severe background slowing with focal or multifocal epileptiform discharges.
In addition to seizures, movement disorders such as ataxia and choreoathetosis are common. Hypotonia, hypertonia, and/or dystonia are present in 50% of cases. In some cases, the patients cannot sit or walk unassisted. Many patients have very little to no speech, and some patients gradually lose eye contact during the course of the disease. SUDEP (sudden unexpected death in epilepsy) has been reported in 12% of cases.
Genotype
Genetics. In four large screens of individuals with epileptic encephalopathies, de novo mutations of SCN8A were identified in 1% of cases (13/1557). The known mutations are all missense mutations except for one splice site mutation that results in an in-frame deletion. Protein truncation mutations have not been observed in EIEE13. In most cases, the mutation arose de novo in the patient. In three cases, the mutation was inherited from a mosaic parent who was unaffected. Approximately 30% of mutations are recurrent. EIEE13 is a dominant disorder, thus affected individuals are heterozygous for the SCN8A mutation.
Most pathogenic mutations of SCN8A are located in the transmembrane domains of the affected protein, as explained below. Details of all the known pathogenic variants will be available soon in an online SCN8A mutation database (see Community below).
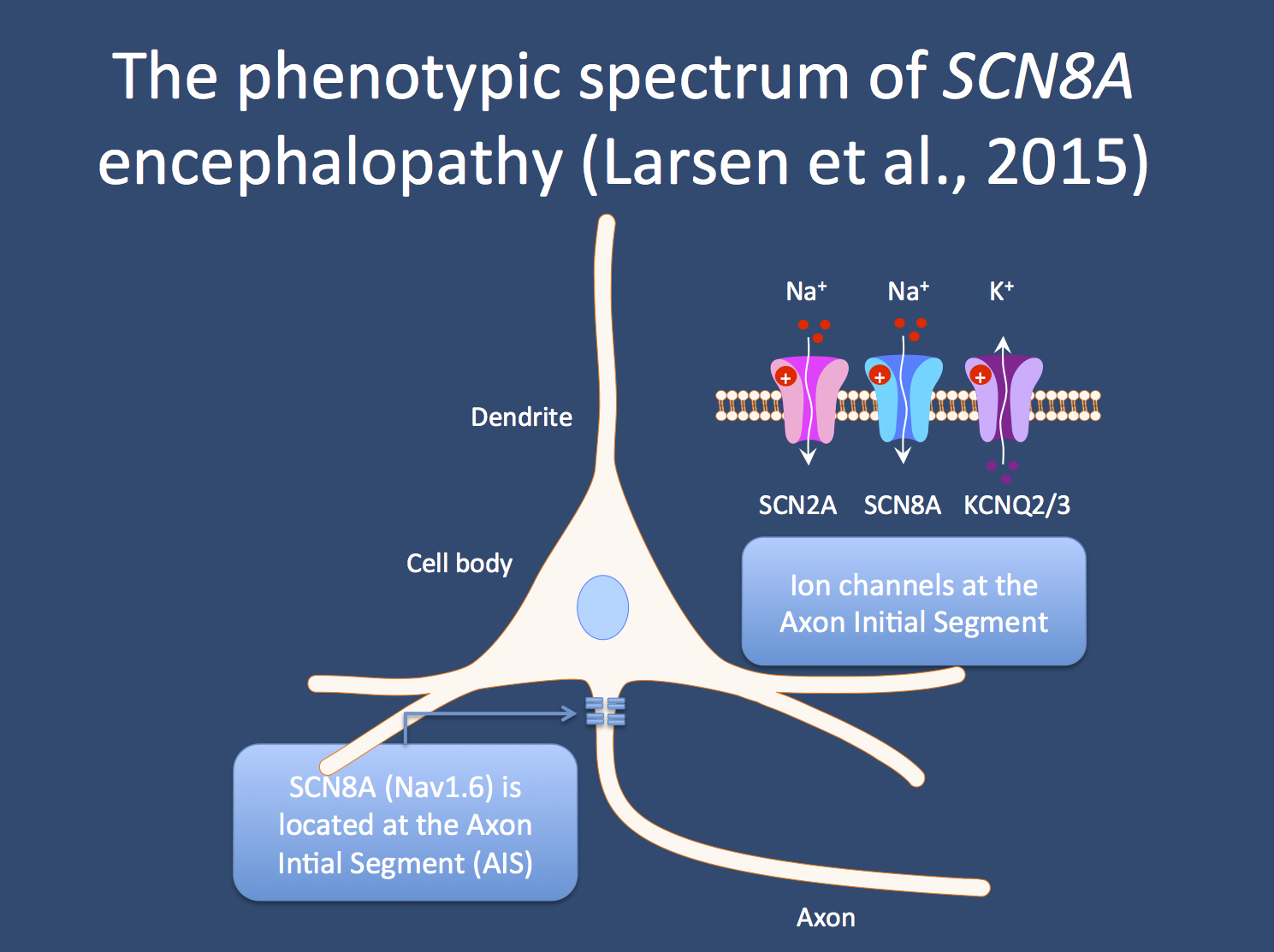
The ion channels encoded by the SCN2A, SCN8A, and KCNQ2 genes are located at the axon initial segment, the part of the neuron where all excitatory and inhibitory impulses at the neuronal membrane are integrated and translated into an action potential. Mutations in all three genes are now associated with severe epilepsies. For SCN2A and SCN8A, the mutations are assumed to be excitatory; for KCNQ2, the mutations are thought to be dominant negative.
Mechanism
Nav1.6. SCN8A encodes the voltage-gated sodium channel alpha subunit Nav1.6. The Nav1.6 protein comprises 4 homologous domains that each have 6 transmembrane segments, two large intracellular loops, the inactivation gate located between D3 and D4, and intracellular N- and C-terminal domains (See Figure). The pathogenic mutations are predominantly located in the transmembrane domains, the inactivation gate, and the C-terminus. These regions of the channel are much more highly conserved through evolution compared to the less well-conserved large intracellular loops and are more likely to be involved in channel function.
Neuronal voltage-gated sodium channels regulate cellular excitability by controlling the flow of sodium ions across the cell membrane. Nav1.6 is expressed in excitatory and inhibitory neurons in the brain. Nav1.6 is localized in high concentrations at the axon initial segments of neurons and at nodes of Ranvier where it mediates action potential initiation and propagation.
Hyperexcitability. Nine EIEE13 mutations have been tested functionally. Seven of the nine demonstrate gain-of-function (GOF) features, including hyperpolarizing shifts in the voltage-dependence of activation, impaired inactivation, and elevated persistent current. These GOF features cause the Nav1.6 channel to be hyperactive, leading to increased neuronal firing. Thus, it appears that neuronal hyperexcitability caused by GOF mutations of SCN8A is the predominant mechanism underlying epilepsy in EIEE13.
Different from Dravet. This mechanism is distinct from the mechanism underlying Dravet syndrome, a key differential diagnosis for EIEE13. More than 85% of Dravet syndrome cases are caused by mutations in the related sodium channel SCN1A. The majority of SCN1A mutations in Dravet syndrome cause loss-of-function of the sodium channel Nav1.1. Thus, medications that further reduce sodium channel activity (e.g. carbamazepine, lamotrigine) are contraindicated in Dravet syndrome. However, in EIEE13, these medications can reduce or prevent seizures in some patients because they may reduce the elevated sodium channel activity observed with EIEE13 mutations. Recently, three loss-of-function (LOF) mutations of SCN8A have been associated with epilepsy. Two missense mutations caused protein instability or loss of channel activity and were associated with EIEE, and one deletion of exons 2-14 was found in a patient with early-onset absence epilepsy.
Community
Community. Researchers and family-run organizations are working closely to share information and develop resources for those interested in EIEE13. A website under development by Dr. Michael Hammer at the University of Arizona, www.Scn8a.net, will serve as a registry for SCN8A families and curate genotype and phenotype information for clinicians and researchers. SPEAK (SCN8A partnership for Epilepsy Awareness and Knowledge) is a collaboration of three non-profit patient advocacy groups: Wishes for Elliott (www.wishesforelliott.org), SCN8A-Help Adeline Find Answers (on Facebook), and The Cute Syndrome Foundation (www.thecutesyndrome.com). The SPEAK partners provide resources for families and raise funds for research on EIEE13.