
Duality. Earlier this week, our Luxembourg collaborators came to visit us at CHOP to discuss our current and future projects. We discussed potential overlaps between the diseases that both our groups are mainly involved in, namely Parkinson’s disease and genetic epilepsies. In fact, we had just published on one of the overlapping genes recently, a gene that we accidentally stumbled upon through our genome sequencing projects. Here is the story of SYNJ1, a gene involved in neurodegenerative phenotypes that link early-onset Parkinson’s disease and epileptic encephalopathy.
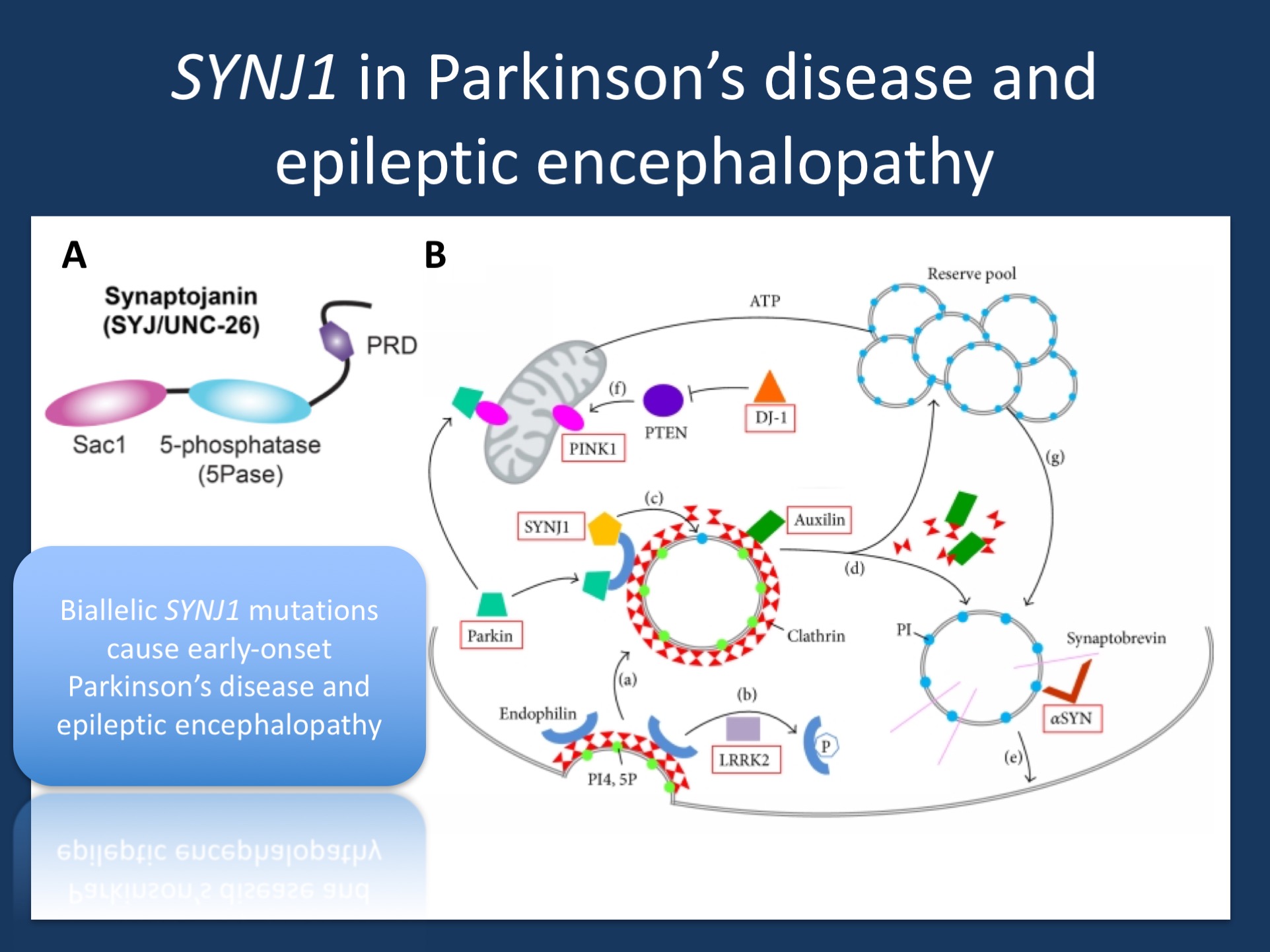
(A) Domain structure of synaptojanin UNC-26. Synaptojanin contains three functional modules: a Sac1 phosphatase domain, a 5-phosphatase domain (5Pase), and a proline-rich domain (PRD). Figure was adapted under a Creative Commons Attribution from Dong and collaborators https://elifesciences.org/content/4/e05660 (B) Synaptic vesicle recycling. (a) During endocytosis, invagination of the clathrin-coated membrane requires endophilin. (b) LRRK2 phosphorylates endophilin, (c) SYNJ1 dephosphorylates PI4,5P into PI, shedding clathrin and its adaptor from the bilayer. (d) Uncoating of the vesicles equires auxilin prior to returning to the reserve pool. (e) Synaptic vesicles are docked and then fused to the membrane by means of a multiprotein complex including synaptobrevin and αSYN. (f) PTEN can increase levels of the PINK1 protein. (g) Proper mitochondrial functioning leads to ATP synthesis, necessary to mobilize the reserve pool of vesicles. PI4,5P: phosphatidylinositol 4,5-bisphosphates; PI: phosphatidylinositol; ATP: adenosine triphosphate; SYNJ1: synaptojanin 1; LRRK2: leucine-rich repeat serine/threonine-protein kinase 2; PTEN: phosphatase and tensin homologue; PINK1: PTEN induced putative kinase 1; DJ-1: Parkinson’s disease protein 7; αSYN: alpha-synuclein. (adapted from Drouet and Lesage under a Creative Commons Attribution http://www.hindawi.com/journals/bmri/2014/289728/).
Opportunity space. Rudi and Roland are part of the LSCB in Luxembourg, the European data hub that we heavily relied on during our EuroEPINOMICS project. Now, within the framework of our emerging center for data-driven discovery in biomedicine (D3b), we invited them over to brainstorm ways to collaborate and – amongst other projects – to connect research on the various disease entities that were involved in, including Parkinson’s disease (LCSB), brain tumor (D3b/CBTTC), and genetic epilepsies (us). It may sound artificial to make these connections at first, but you end up shifting perspective by forcing yourself to think across disease domains. You are creating an “opportunity space” where you have the chance to seek out novel hypotheses across domains. The overlap between epileptic encephalopathies and brain tumors? mTOR pathways, various cancer genes remerging as EE genes through de novo mutations. The overlap between epileptic encephalopathies and Parkinson’s? Amongst other genes, synaptojanin 1, encoded by the SYNJ1 gene. Here is the story of a protein that has its two-faced nature deeply embedded in its name.
The anatomy of SYNJ1. The synaptojanin 1 protein derives its names from two parts, the synapse and Janus, the roman two-faced god of beginnings, gates, transitions, and time. The janus aspect of synaptojanin 1 refers to the fact that the protein contains two subsequent functional domains that have phosphatase activity. In technical terms, both domains are referred to as an N-terminal Sac1-like inositol domain (Sac1) and a central 5’-phosphatase domain (5’PP). Synaptojanins are involved in dephosphorylation of phosphatidylinositols, a phosphoinositide highly concentrated at the plasma membrane. Both domains help break down phosphatidylinositols, which is important in getting rid of the clathrin coat that surrounds vesicles undergoing endocytosis. Basically, a synaptic vesicle that is recycled from the plasma membrane is surrounded by a clathrin coat. This coat is necessary to accomplish endocytosis at all, as it is required for the formation of budding vesicles. However, during the further steps of vesicle recycling, this coat is in the way and needs to be removed. This is where synaptojanin steps in.
The genetics or SYNJ1. Bi-allelic mutations in SYNJ1 have been associated with two different disease phenotypes, early-onset Parkinson’s disease and a severe early-onset epileptic encephalopathy with neurodegenerative features. At first, both phenotypes have little in common. The movement disorder phenotype usually starts in the 30’s with predominant features of Parkinsonism. In contrast, the epileptic encephalopathy phenotype start in the first year of life with seizures as the most prominent feature. In our recent publication by Hardies and collaborators, we investigated the genetic basis of three families with early onset refractory seizures and progressive neurological decline. All three families underwent exome and genome sequencing and were included in this project because bi-allelic pathogenic variants in SYNJ1 had been identified. In our current publication, we describe the phenotypes and investigate the functional consequences of SYNJ1 dysfunction.
Missense or knock-out. All three bi-allelic mutations in the early-onset epilepsy families were either truncating mutations, leading to decreased mRNA levels, or missense mutations that lead to a profound impact on synaptojanin 1 function. Interestingly, the homozygous p.Tyr888Cys variant identified in Family A was in the 5’PP domain. Even though this substitution only affected the second domain, it led to a decreased activity of both the proximal Sac1 domain and distal 5’PP domain. In brief, even though this variant only led to a substitution in the second domain, the functional consequence was so severe that the synaptojanin 1 protein was virtually non-functional. This is in contrast to the p.Arg258Gln substitution that was found in families with early-onset Parkinson’s disease that exclusively affected the function of the Sac1 domain while leaving 5’PP domain function unaffected. Accordingly, the functional studies in the publication by Hardies and collaborators helped understand why SYNJ1 pathogenic variants can result in two different phenotypes that may be the extremes of an underlying phenotypic spectrum – the pathogenic variants identified in families with early-onset Parkinsonism leave some of the dephosphorylation activity of synaptojanin 1 intact, while pathogenic variants in families with early-onset epileptic encephalopathies result in a complete loss of synaptojanin 1 function, either through truncating mutations or missense variants that result in an impairment of both domains.
What you need to know. SYNJ1 encodes for synaptojanin 1, a protein critically involved in the processing of recycled synaptic vesicles. Biallelic pathogenic variants in SYNJ1 can result in two phenotypes, including familial Parkinson’s disease starting in early adulthood and severe early-onset epileptic encephalopathy with neurodegenerative features starting in infancy. The severity of the phenotype is correlated with the degree of impairment of both phosphatase domains of the synaptojanin 1 protein.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
