
Inhibition. GABA is the main inhibitory neurotransmitter in the the Central Nervous System. Given that epilepsy is typically associated with increased excitability, all mechanisms related to GABA signaling are of natural interest to the epilepsy community. Almost 15 years ago, mutations in GABRA1, coding for alpha-1 subunit of the GABA-A receptor, have been identified in familial Juvenile Myoclonic Epilepsy, but there has been relative silence around this gene since. Now, two publications highlight the other side of GABRA1 as a gene for epileptic encephalopathies, putting the GABA receptor into the spotlight again.
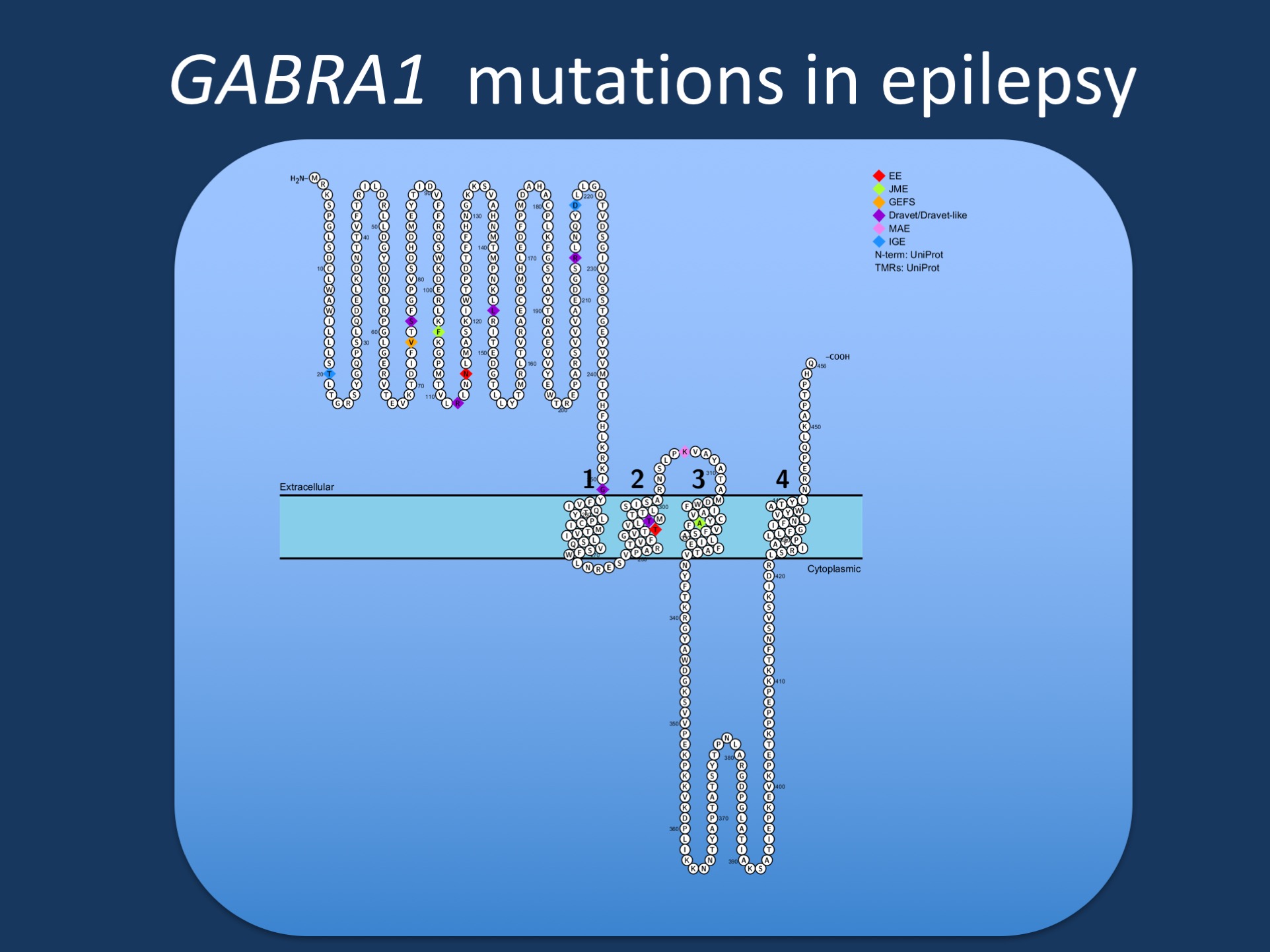
Figure. Pathogenic variants in GABRA1 in various phenotypes reported by Johannsen and collaborators. GABRA1 was initially identified in patients with Juvenile Myoclonic Epilepsy (JME), but most reported patients to date have more severe epilepsies such as epileptic encephalopathies (EE), Juvenile Myoclonic Epilepsy (JME), Dravet Syndrome, or Myoclonic Astatic Epilepsy (MAE). Few patients have milder phenotypes such as Genetic/Generalized Epilepsy with Febrile Seizures Plus (GEFS+).
GABA. Whenever I think about GABA, I recall a lecture in medical school when once of my professors called gamma-amino-buteric acid the “main transmitter in the brain that says NO to neurons”. The functioning of our neuronal networks is critically dependent on the proper interaction between excitation and inhibition and the inhibitory side, mainly involving GABA signaling, plays by its own rules. The GABA-A receptor is composed of five subunits, including two alpha subunits that are typically encoded by the GABRA1 gene. The other subunits are beta subunits and gamma subunits. The latter are encoded by GABRG2, which is also associated with familial idiopathic/genetic generalized epilepsies. In summary, genes for GABA receptors are a natural candidate.
GABRA1. Initially, GABRA1 was identified as the causative gene for a familial Juvenile Myoclonic Epilepsy (JME) in a large French-Canadian family. This family carried a p.A322D pathogenic variant in GABRA1. After this, several individual reports have highlighted de novo GABRA1 mutations in patients with absence epilepsy and more severe epilepsies such as Dravet Syndrome. However, a systematic analysis of how pathogenic variants in GABRA1 manifest themselves phenotypically, was missing. Now, two publications highlight the emerging spectrum of GABRA1 mutations. In a recent publication in Epilepsia, Kodera and collaborators identify de novo pathogenic variants in GABRA1 in patients with Infantile Spasms and Ohtahara Syndrome. At the same time, we are part of a publication by Johannsen and collaborators in Neurology, which highlights the phenotypic spectrum of 17 additional individuals carrying pathogenic GABRA1 variants. So what do these two publications tell us about GABRA1?
The phenotypic spectrum. Even though Juvenile Myoclonic Epilepsy was the first GABRA1 phentoype to be identified, it appears to the least prominent phenotype in 2016. Since the initial report by Cossette and collaborators, less than five patients with JME carrying pathogenic GABRA1 variants have been identified, while the majority of patients have epileptic encephalopathy. There is a huge bias in this assessment, given that most patients with non-familial mild epilepsies do not undergo genetic testing. However, I think it is safe to say that GABRA1 is not a prominent JME gene given the data available to the consortia who have worked on the genetics of the idiopathic/genetic generalized epilepsies. The more severe GABRA1 phenotypes include patients with Ohtahara Syndrome, Infantile Spasms, Myoclonic Astatic Epilepsy (Doose Syndrome), and other unclassified epileptic encephalopathies. The pathogenic variants are thought to results in haploinsufficiency and there is no obvious genotype/phenotype correlation
The unifying theme. Categorizing patients into diagnostic entities sometimes blurs the perspective for shared phenotypic features. Accordingly, the study by Johannsen and collaborators, we also looked at specific seizure types, EEG patterns, and other phenotypic features. It is interesting to note that generalized spike-wave activity is observed in 90% of all patients and up to 50% of patients have a photoparoxysmal response. These features are typically associated with generalized epilepsies and are somewhat of an uncommon finding in early-onset epileptic encephalopathies. Taken together, the GABRA1 phenotypic spectrum is unified by EEG features reminiscent of generalized epilepsies, even in patients with phenotypes where these features are not very common.
This is what you need to know. Two recent publications highlight the phenotypic spectrum of pathogenic GABRA1 variants in human epilepsy. Initially considered a gene for Juvenile Myoclonic Epilepsy, GABRA1 is re-emerging as a gene for epileptic encephalopathies with a broad phenotypic range. Some of the de novo pathogenic variants seem to be recurrent, including p.R112Q, p.G251D, p.M263T, and p.K306T/I, which may help identify further cases in the absence of segregation data. Haploinsufficiency is considered the pathogenic mechanism.
Epilepsiome page. Katrine Johannsen, the first author of the Neurology GABRA1 publication, has compiled the Epilepsiome page for GABRA1, which can be found here. As with the other Epilepsiome pages, it will go through a regular review and we will let you know about the updates.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.

If haploinsufficiency is the mechanism, we would expect to see frameshift and nonsense mutations as approximately half of the total – is that the case for GABRA1?
Hi Miriam,
this is actually a good question that I was thinking about when writing this, as well. At this point, the number of patients is probably too small to precisely quantify the fraction of patients with truncating mutations. However, the relative frequency of recurrent mutations is intriguing, which would argue against a simple haploinsufficiency mechanism.