
Recessive epilepsies. Dravet Syndrome is one of the most prominent genetic epilepsies and presents in the first year of life with prolonged fever-associated seizures. Haploinsufficiency of SCN1A, either through mutations or deletions, is the major cause of Dravet Syndrome. In a recent publication in the European Journal of Pediatric Neurology, two families with recessive Dravet Syndrome and biallelic SCN1A variants are reported. Let’s have a look at how to interpret these findings.
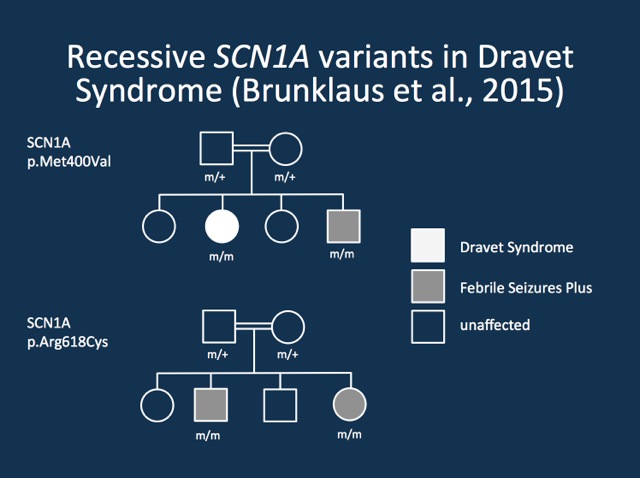
Cause or coincidence. Two recessive families with Dravet Syndrome/GEFS+ and recessive mutations in SCN1A reported by Brunklaus and collaborators.
De novo mutations. Dravet Syndrome is caused by de novo alterations in the SCN1A gene in more than 80% of patients. Even in patients with reportedly negative SCN1A testing, some patients may have SCN1A mutation at second glance. This very strong association suggests that genetic alterations in SCN1A, which result in haploinsufficiency, are the major cause for Dravet Syndrome. Haploinsufficiency refers to the situation when only a single functional copy of a gene is present and the gene product of only one copy is insufficient. In addition to SCN1A, other genes have been implicated in Dravet Syndrome, such as GABRA1 and STXBP1. However, the close connection between Dravet Syndrome and SCN1A is one of the strongest genotype/phenotype correlations that we know in neurogenetics today. The publication by Brunklaus and collaborators raises the issue whether this correlation is sufficiently strong to also implicate genetic variants in SCN1A other than de novo mutations in Dravet Syndrome. In other words, just because we know that de novo mutations in SCN1A cause Dravet Syndrome, would we automatically assume that any other inherited rare variation in SCN1A must be causative? Would our level of suspicion by sufficiently high not to pursue other genetic investigations if we find recessive variants?
Family studies. Brunklaus and collaborators report on two consanguineous families of South Asian origin. Each family has two affected children with Dravet syndrome and fever-associated epilepsies within the GEFS+ spectrum. Given the Dravet-like presentation, the authors specifically analyzed the SCN1A gene – in both families they identified novel SCN1A variants that were homozygous in both affected sibs and heterozygous in parents. The SCN1A variants in both families are not observed in the ExAC database, the largest exome database to date. Even though the authors do not mention the transcript they are referring to, it probably refers to the transcript that is also used in the ExAC browser, at least by comparison of the amino acid positions mentioned in the publication.
Functional data. In their current study, the authors do not provide functional data that support their claim that the two novel SCN1A mutations are causative, and it will be interesting to see future functional work on both variants. The authors speculate that both mutations may exert a small effect that is insufficient when heterozygous, but cause Dravet Syndrome when present in a homozygous state. They refer to the studies from SCN1A mouse models where such scenario has been observed.
Cause or coincidence? The study by Brunklaus and collaborators raises the following question: What is more likely? The fact that both variants are causative versus the fact that both variants are coincidental? The tight connection of SCN1A with Dravet Syndrome is intriguing. However, the authors were unable to provide segregation data in unaffected siblings. Additionally, the fact that both families are from a population that is underreported in genetic databases may suggest that both variants are benign population variants that are non-causative. In either case, a full genetic work-up through exome sequencing including assessment for possible de novo mutations that might be present in a mosaic state in parents would be interesting. If these investigations would be negative and if functional data points towards an effect of these variants on channel function, there would be strong argument to consider the recessive SCN1A mutations as causative.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.

My daughter has dravets syndrome but her gene I’d named pcdh19 there was no mention of that gene in this article what can you tell me about dravets and her gene?
Dear Jennifer,
apologies for the delay in responding – yes, we have left our PCDH19. There is a long debate on whether the clinical picture of girl with epilepsies due to PCDH19 mutations should be classified as Dravet Syndrome or whether it is a distinct disease that is different from it. Depending on who you ask, you will get different views on that. This is the main reason why we usually don’t include PCDH19 in our list of Dravet genes. However, you raise an important point: we have written very little about PCDH19 in the last two year and we’ll try to improve this!