
Rare human knockouts. Recessive disorders arise when both copies of a causative gene are affected by mutations. These diseases are thought to be a very rare occurrence, but the cumulative impact of these conditions is not known. Population genome sequencing offers the possibility to assess the spectrum and distribution of potentially causative mutations in large groups of individuals. In a recent publication from deCODE published in Nature Genetics, the authors examine the population spectrum of rare human knockouts using the unique genetic data and population structure of the Icelanders. Here is the story about potential candidate genes identified by population genetics.
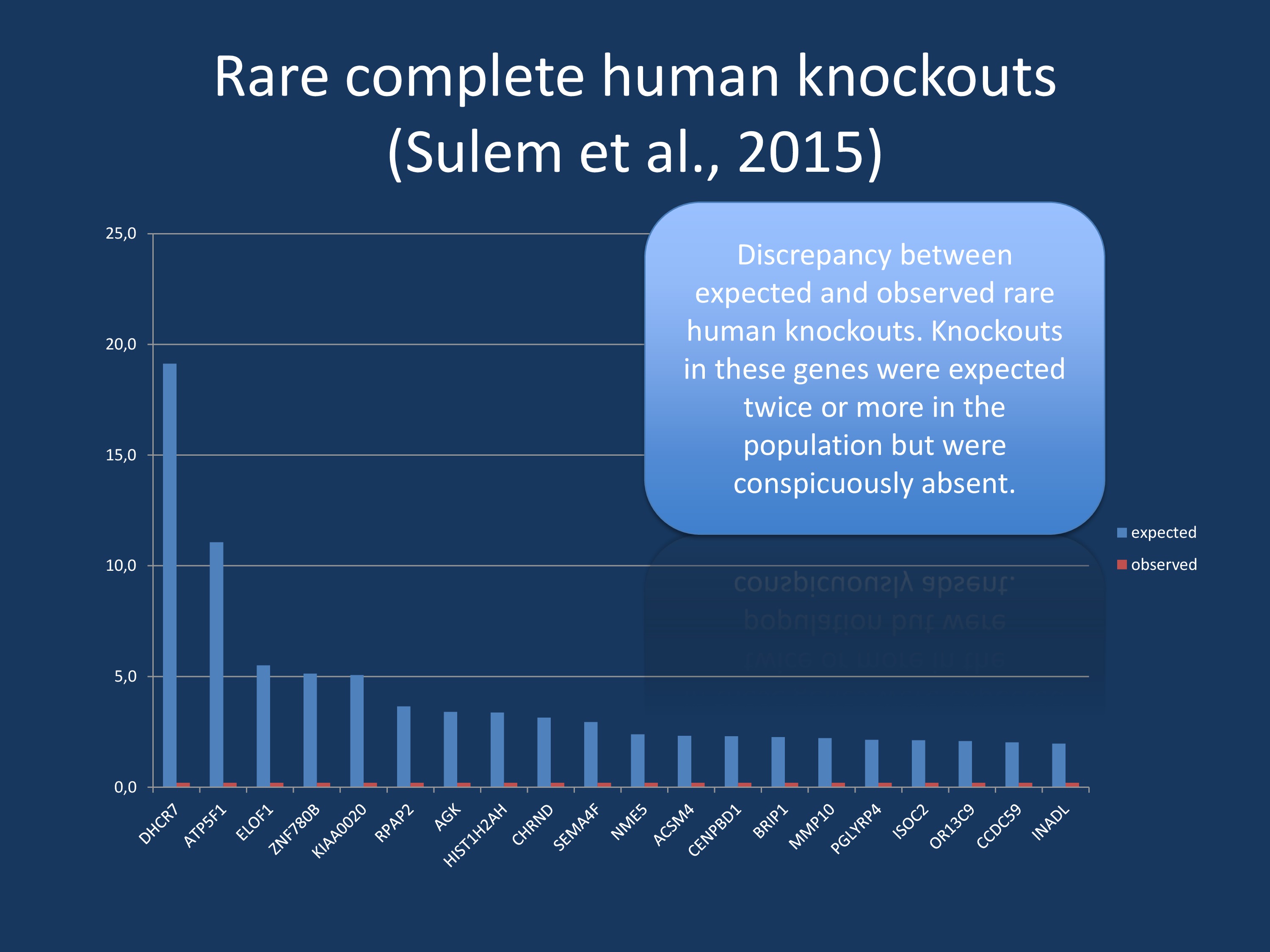
Data derived from Supplementary Table 4 by the publication by Sulem and collaborators. We have filtered their data for variants that are expected in two or more individuals, but were not observed in anyone. Interestingly, there are a few known disease genes intermixed in this list of genes including DHCR7 and CHRND. Analyses like this may help identify novel recessive disorders, and, in the case of genes with known homozygotes, the phenotyping of mutation carriers.
100K genotypes. The method behind the recent publication by Sulem and collaborators can be explained in a few sentences. First, the authors sequenced roughly 2600 individuals from Iceland on a genome-wide level. Secondly, they identified rare, deleterious variants in these individuals. Third, given the unique population structure in Iceland, they were able to extrapolate the frequency and combination of these variants in a larger population of more than 100,000 individuals that were genotyped with low-cost SNP arrays. Particularly the step from knowing the sequence in 2600 individuals to expanding this to more than 100,000 individuals is the real “magic” of this publication that is made possible by a method called imputation.
Imputation. Imputation refers to the extrapolation of genotypes at a given location in an individual based on surrounding genetic data. For example, if we know that a certain combination of common Single Nucleotide Polymorphisms (SNPs) is associated with a loss-of-function mutation in an individual who was genome-sequenced, we might also expect the same loss-of-function mutation when we encounter the same combination of common SNPs again. This is true provided that the population is not too heterogeneous and that the researchers have done some basic footwork to show that the overall assumptions are true in their population – both are true for the Icelandic population and the genetic screen performed by deCODE. Basically, in Icelanders, if you identify a single rare disruptive variant, you can easily assess the frequency and combination of homozygotes and heterozygotes in a very large population. And you can ask the question what happens to patients who are homozygous.
Rare variants. Sulem and collaborators were interested in identifying potentially recessive disorders by genotyping first. In the genome-sequenced patients, they identified roughly 5000 genes that carry rare disruptive mutations that were present in less than 2% of individuals on a population level. For 1171 of these genes, they found that a certain proportion of Icelanders are either homozygous or compound heterozygous. The cumulative frequency of all homozygous and compound heterozygous mutation carriers was roughly 7.7%. The next task that Sulem and collaborators aimed to do was to phenotype these human knockouts and assess whether they have medical conditions that may be attributed to these gene knockouts. As a first, step, they were able to link their genetic data with death records in the Icelandic population, for example children who died before the age of 15. This may be an indication that there might have been severe genetic disorder present in these individuals.
Examples. The data provided by Sulem and collaborators allows us to filter for genes where the highest age of homozygotes was 15 years or less. This leads to the BRF2 gene, a gene not involved in human disorders yet. Looking at the data a bit closer, we find that within the genotyped population, we would have expected ~7 homozygotes for the disruptive BRF2 mutation. However, only a single individual was observed. This deviation of the observed from the expected number of individuals is another hint that this gene may be disease-related (deviation from Hardy-Weinberg equilibrium). Again, the next step will be to phenotype these homozygous individuals and possibly connect this with a disease. Another look at the freely available data by Sulem and collaborators allows us to select for genes that are NEVER seen as homozygotes even though we would expect several individuals.
Zero homozygotes. There a few very drastic examples of rare, disruptive mutations that are expected in several individuals, but are simply not observed in the population. Most prominently, a disruptive mutation in DHCR7 is expected in almost 20 individuals, but never observed in anyone. It is known that recessive mutations in this gene cause Smith-Lemli-Optiz syndrome, an autosomal recessive multiple congenital malformation and intellectual disability syndrome. Another gene, CHRND coding for the delta peptide of the nicotinergic acetylcholine receptor, is expected in 3 individuals, but also never observed. Mutations in this gene are known to cause the lethal form of multiple pterygium syndrome (LMPS), a lethal neuromuscular syndrome. In summary, the zero homozygote analysis already identified several severe genetic diseases and may suggest novel genes that cause phenotypes so severe that they are incompatible with life.
This is what you need to know. The study by Sulem and collaborators provides the basis for a “reverse genetic approach” to recessive disorders by providing a genetic population map of rare human knockouts, which are prime candidates for novel diseases. This approach differs from the classical way of looking at recessive disease by identifying families and identifying the corresponding genotype. It will be interesting to see whether some of the phenotypes may result in particular neurological disorders, which may help decipher the genetic architecture of these conditions.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
