
Electrolytes. Sodium, calcium, and magnesium – I usually tell my students that imbalances in these serum electrolytes may result in seizures, when levels fall under a critical threshold. Amongst these imbalances, hypomagnesemia, a reduction of the serum magnesium level below 0.7 mmol/L, is a very rare cause of seizures, particularly in a pediatric population. However, there are genetic conditions that result in reduced magnesium levels and lead to neurological complications. In a recent paper in PLOS Genetics, the phenotype of CNNM2 mutation carriers is investigated – and magnesium is only the beginning of the story.
CNNM2. Let’s start with the kidneys, organ of the body that appear to be entirely unrelated to epilepsy in the first place. In order to retain magnesium in the serum, the kidneys need to reabsorb a significant fraction of the excreted magnesium in the distal convoluted tubule (DCT). For this reabsorption, magnesium needs to be actively transported from the urine back into the bloodstream, a process that requires coordinated uptake by the lining cells (apical side), transport through the cell, and extraction into the blood vessels (basolateral side). While the apical transport is well characterized and accomplished by TRPM6 and TRPM7 channels, the basolateral mechanisms are much less understood. However, CNNM2 coding for cyclin M2, seems to play a major role in intracellular magnesium sensing in the DCT, which is necessary for proper release of magnesium into the bloodstream. If CNNM2 function is impaired, the kidney is losing magnesium, which leads to hypomagnesemia and neurological complications in mutation carriers – but there is much more it.
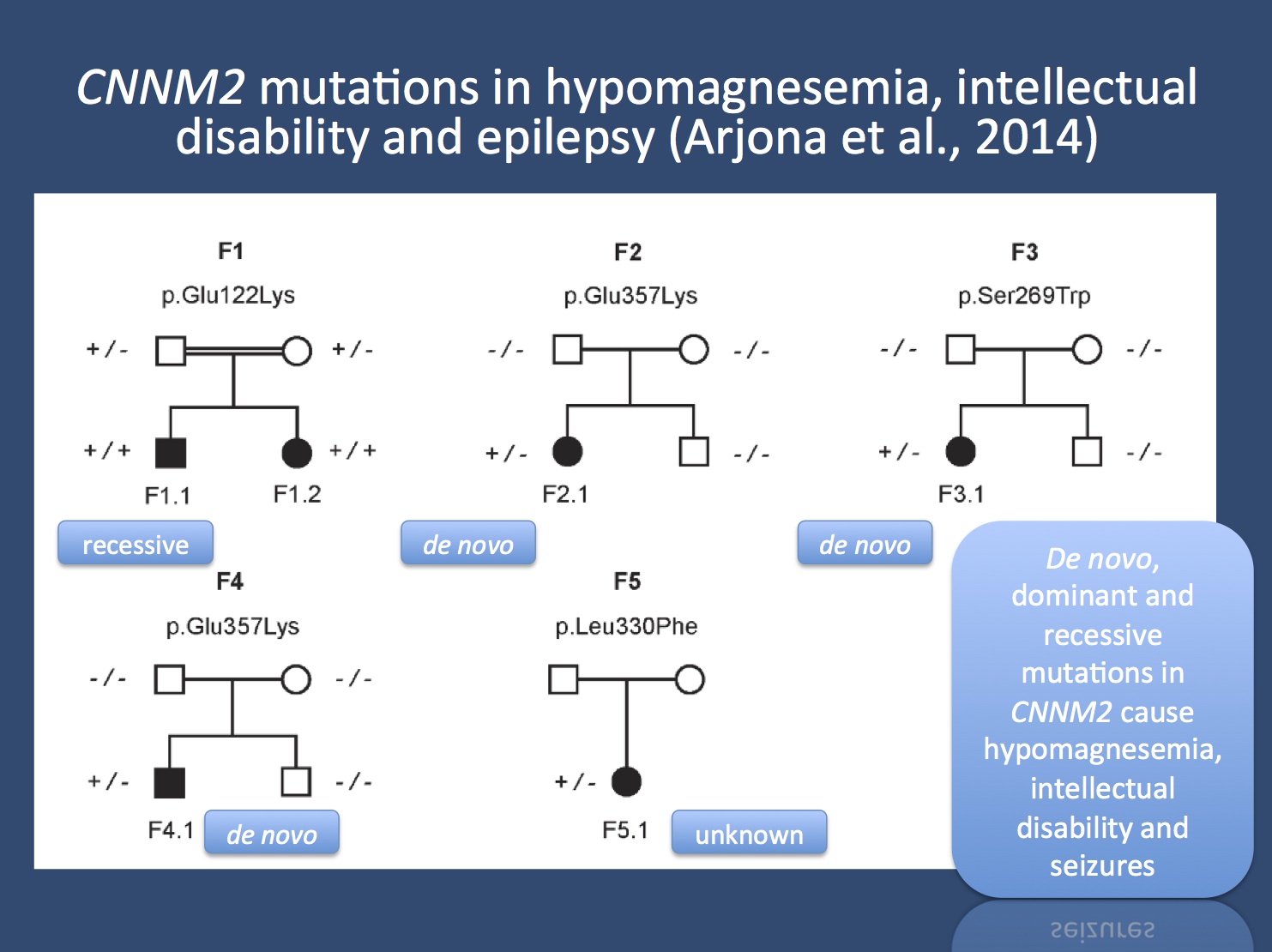
Pedigrees of the families with mutations in CNNM2. Image reproduces under a Creative Commons License from www.plosgenetics.org/article/info%3Adoi%2F10.1371%2Fjournal.pgen.1004267
Mutations. Arjona and collaborators screened patients with hypomagnesemia, intellectual disability, and epilepsy. In 2011, their group had described two small families with dominantly inherited hypomagnesemia and CNNM2 mutations. In their current publication, they extend their mutation search and identify one family with a recessive mutation, three patients with de novo mutations, and one patient with a CNNM2 mutation and unknown inheritance. The common phenotypic feature was the magnesemia, but all patients also had epilepsy and various degrees of intellectual disability. Seizures were usually not responsive to magnesium supplementation, suggesting that there is more to the story than the serum magnesium level. In fact, CNNM2 is also important in brain development.
CNS phenotypes. The authors assessed the role of CNNM2 gene ablation in zebrafish and found that zebrafish larvae have an overall reduction in brain size – a phenomenon also seen in some patients with CNNM2 mutations. Interestingly, this was independent of magnesium normalization in zebrafish, suggesting profound effects of CNNM2 on brain development independent of magnesium levels. In fact, CNNM2 might have entirely independent functions in the brain.
Inheritance. It is interesting to see that dominant, de novo, and recessive mutations in CNNM2 can lead to similar phenotypes including hypomagnesemia, intellectual disability, and seizures, a pattern that is only seen in few epilepsy-related genes. Given that serum magnesium serves as a biomarker for this condition, the identified mutations are credible. However, the magnesium levels might be unrelated to the patients’ intellectual disability and epilepsy.
Lessons. CNNM2 mutations have not been identified in the Epi4K study or other exome studies in patients with autism, intellectual disability, and schizophrenia. Accordingly, CNNM2 mutations may in fact represent a distinct phenotype that is not easily confused with other epileptic encephalopathies. While the effects of CNNM2 mutations on brain development remain to be explored, low magnesium seems to be a reliable biomarker for this disease. Finding hypomagnesemia in a patient with a neurodevelopmental disorder may prompt analysis of this gene.
Child Neurology Fellow and epilepsy genetics researcher at the Children’s Hospital of Philadelphia (CHOP), USA and Department of Neuropediatrics, Kiel, Germany

