
Climate change. In the era of exome and genome sequencing, it might be worthwhile revisiting the merit of family studies in epilepsy research. Seizure disorders are known to have a highly diverse genetic architecture. When singleton studies identify a single, unique gene finding, this discovery usually does not provide much information about the potential causal role of the variant given the high degree of genomic noise. In contrast, family studies are usually considered more robust, as segregation of variants can be traced. Here is the inconvenient truth: unless the family is very large, segregation of possibly monogenic variants adds little information given the vast amount of variants present in our genomes.
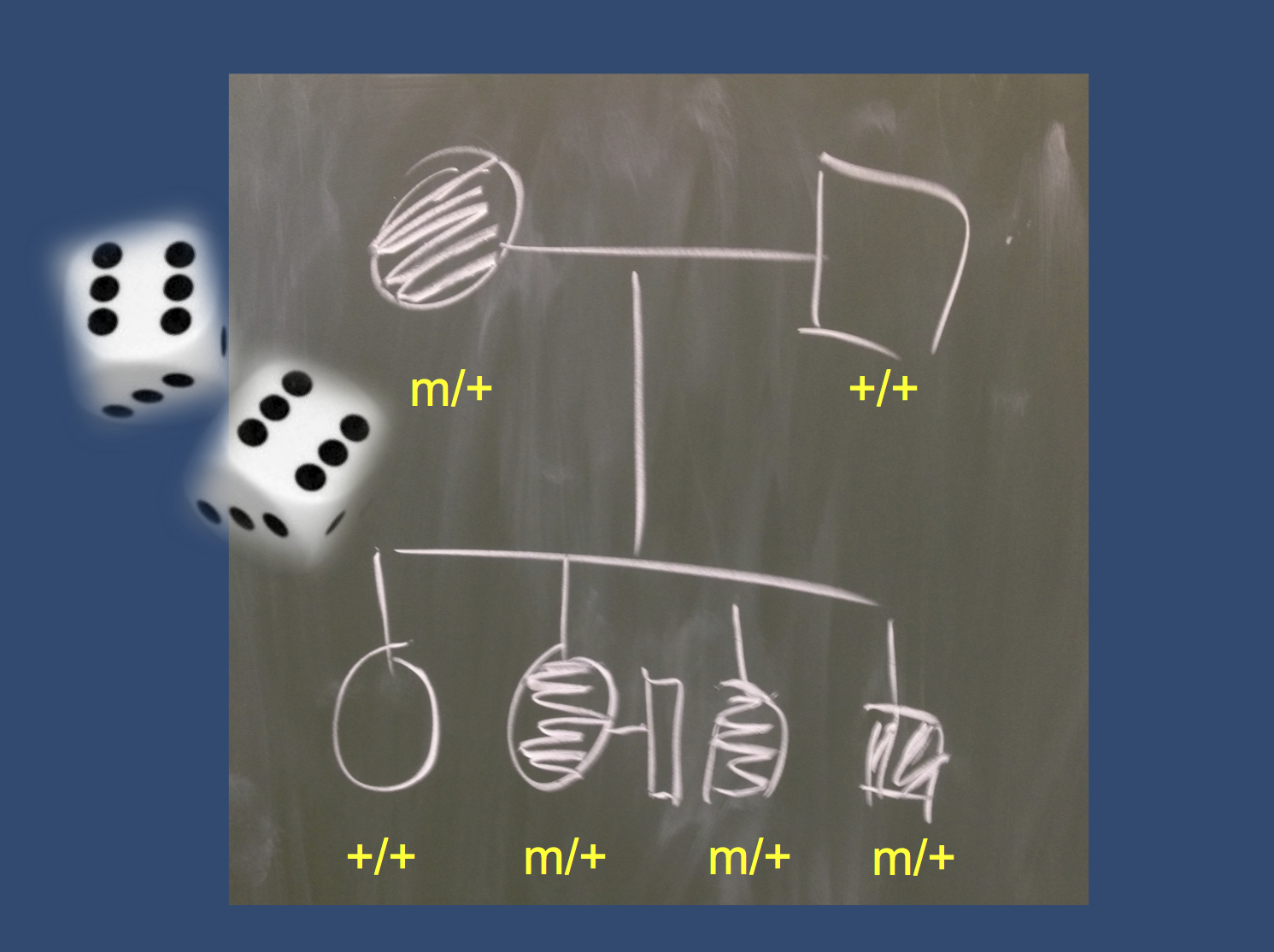
An example pedigree (photograph taken at the EuroEPINOMICS General Assembly in Tübingen). The constellation of a variant completely segregating with the disease is a matter of how many variants were tested. If only a single variant was analyzed, the probability is already twice as high as rolling two “sixes” simultaneously. When more than one variant is tested, this probability may even rise to more than 95% in this family.
Clinical encounter. Some forms of generalized epilepsies may run in families in an autosomal dominant manner including monogenic forms of Genetic Epilepsy with Febrile Seizures Plus (GEFS+) or Idiopathic/Genetic Generalized Epilepsies (IGE/GGE). Most patients with epilepsy, however, do not have a positive family history. Accordingly, encountering an epilepsy family with four or five affected individuals is a rarity in clinical practice, even in centers specializing in epilepsy genetics. If such families are seen in clinic, you usually have the feeling that gene identification is within reach, either through candidate studies or hypothesis-free through exome or genome sequencing. Statistically, this might not be further from the truth.
The omics era. Family studies in small families have worked well in the pre-omics era. If a family with four individuals affected by GEFS+ is found to have a mutation in SCN1A that segregated perfectly, there has been little debate whether this variant was considered causal. SCN1A is the major gene for GEFS+, and complete segregation with the phenotype in four affected individuals is possibly even more than you would expect from SCN1A given the incomplete penetrance observed in some families. SCN1A is a well-established gene for monogenic epilepsies, and we know about the associated phenotypes. However, problems will arise when novel genes are searched for using omics technologies.
Running in the family. The lack of discriminatory power to tell pathogenic variants from randomly co-segregating variants can be illustrated by the following back-of-the-napkin calculation: imagine a family with four individuals affected by a disease inherited in an autosomal dominant manner (Figure). The probability that any variant identified in the affected parent segregated perfectly in the four children (three affected, one unaffected) is 6%. This probability is twice as high as the rolling the number six two times. If we look at more than one variant, which is a scenario that is frequently encountered in exome/genome studies, the probability of having any variant that segregates perfectly might actually get quite high. If 10 variants are followed up, the probability that any variant segregated perfectly is roughly 50%. If 50 variants are followed up, this probability rises to 95%. Accordingly, even though small families may appear promising at first glance, identifying novel genes may be difficult given the massive number of parallel tests. While there are well-establish cut-offs for genome-wide significance levels for family studies (LOD score of 3 or higher), most smaller families will never reach this significance level.
A possible solution. The most straightforward solution to ascribe a pathogenic role to a novel candidate gene is independent replication, i.e. finding a second family with a mutation in the same gene. This however, may never happen given the likely genetic heterogeneity of the epilepsies. Therefore, other possible ways of telling causative variants from harmless genomic variation are necessary. In previous posts, we have discussed scores for mutation intolerance or pathogenicity. These scores and related measures may be used to zero in on a possible candidate variant, and perfect segregation with the phenotype may help generate additional evidence. For these kinds of studies, reference values and reliable significance levels need to be established. And even then, the causative gene in some smaller families may be difficult to pinpoint. We might have difficulties identifying the causative gene that is hidden before our eyes in the river of rare variants.
Child Neurology Fellow and epilepsy genetics researcher at the Children’s Hospital of Philadelphia (CHOP), USA and Department of Neuropediatrics, Kiel, Germany


Pingback: From unaffected to Dravet Syndrome – extreme SCN1A phenotypes in a large GEFS+ family | Beyond the Ion Channel
Pingback: Modifier genes in Dravet Syndrome: where to look and how to find them | Beyond the Ion Channel
Pingback: Mining GWAS mountains for missing heritability | Beyond the Ion Channel
Pingback: The age of mega-genomics, type 2 diabetes, and protective variants in SLC30A8 | Beyond the Ion Channel
Pingback: A polygenic trickle of rare disruptive variants in schizophrenia | Beyond the Ion Channel
Pingback: 9 things you didn’t know about bioinformatics | Beyond the Ion Channel
Pingback: Living in a post-linkage world, craving knowledge | Beyond the Ion Channel