
Success rate. What is in an exome? There are lots of rare and unknown variants that are hard to make sense of unless we can ask a specific question or have further data to limit the number of genes that we look at. Genetic studies in recessive diseases with limited candidate genes to consider might represent one of the most straightforward cases. In a recent paper in BMC Neurology, the genetic cause of a recessive epilepsy/intellectual disability syndrome in a consanguineous family presenting with primary microcephaly was solved using a single exome of an affected individual. Was this just lucky or can this strategy be applied to any recessive family with a reasonable chance?
Microcephaly. If the head circumference of an individual is more than 2 standard deviations below the mean when adjusted for age and gender, this is referred to as microcephaly. Microcephaly can be primary, when present from birth, or secondary, developing during the first years of life. The latter may occur in patients with severe epileptic encephalopathies. Primary microcephaly is sometimes found in families with a recessive inheritance pattern, which is referred to as primary autosomal recessive microcephaly (MCPH, OMIM 251200). As of 2014, up to 10 genes for MCPH are known including ASPM, MCPH1, WDR62, CDK5RAP2, CASC5, CENPJ, STIL, CEP135, CEP152 and ZNF335. Amongst the various genes, ASPM and WDR62 appear to be the most frequent causes of MCPH. The frequency of these mutations in sporadic microcephaly, however, is not known.
Microcephaly and brain development. Malformations of cortical development are usually classified according to the Barkovich classification. In brief, malformations of cortical development may be secondary to abnormal neuronal and glial proliferation or apoptosis (Group 1), due to abnormal neuronal migration (Group 2), or secondary to abnormal postmigrational development (Group 3). This distinction should somehow reflect the different developmental stages and time points at which the malformation occurs. Lissencephaly, for example is due to abnormal migration (Group 2), polymicrogyria is a disorder of cortical organization (Group 3). Microcephaly, in contrast, is due to reduced neuronal proliferation, an event that occurs very early in brain development (Group 1). The WDR62 protein is involved in mitotic spindle formation, an essential component during cell division.
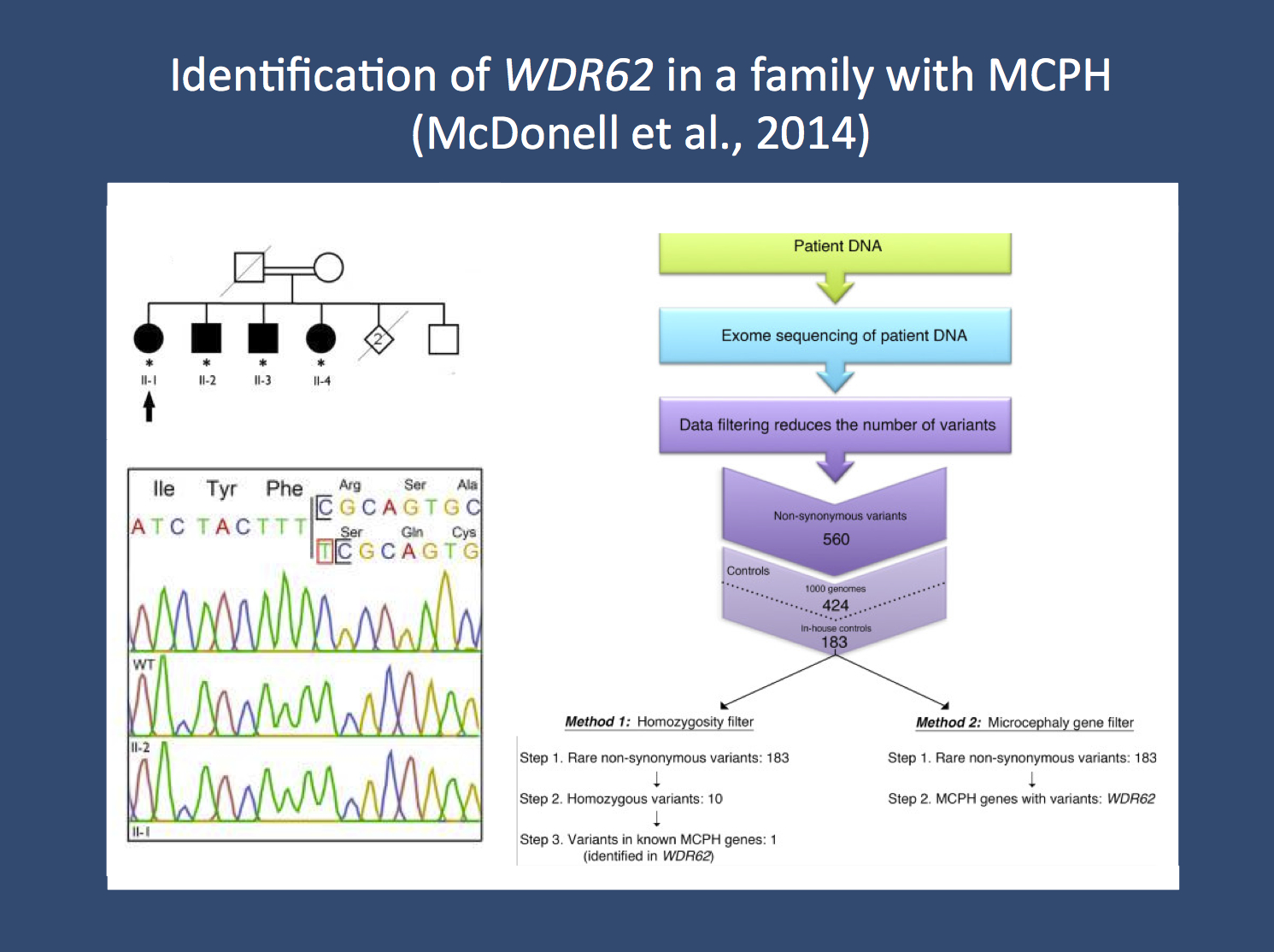
Analysis strategy in the publication by McDonell and collaborators. The researchers looked for the cause of the primary autosomal recessive microcephaly (MCPH) in a family with four affected siblings (upper left corner). They performed exome sequencing in one family member and used stringent filtering to zoom in on the culprit mutation, a homozygous single base pair insertion in WDR62 (right panel). This mutation was confirmed through Sanger sequencing (lower left panel). The figure was adapted from the open access publication by the authors under a creative commons licence. Source: http://www.biomedcentral.com/1471-2377/14/22)
Finding WDR62. McDonell and colleagues were faced with the question of identifying the causative gene in a consanguineous family with microcephaly. Four siblings in their fifties were affected, and the parents were not available. The patient material was furthermore sent in from a remote region in Canada, allowing the researchers little back-and-forth to evaluate patients and look for possible additional clues. If faced with such a situation, what is the best and most cost-effective possibility to identify the culprit gene? The authors point out that a commercially available microcephaly panel would have cost more than 5000 USD. The authors turned towards exome sequencing, filtered the data down using a two-winged approach, looking for a panel of microcephaly genes and recessive variants. Both filtering strategies left them with a single causative gene, a homozygous mutation in WDR62 that could also be confirmed in the other affected siblings.
Linkage no more. Let’s quickly review what the authors did not do. It might have been cost-effective to perform homozygosity mapping in the sibs to identify joint regions that are homozygous in all patients. Alternatively, the authors could have sequenced the two most likely MCPH genes, ASPM and WDR62. It is difficult to tell which strategy would have been cheaper. Also, the authors do not comment on the coverage of the microcephaly genes in the exome data they analyzed. Therefore, there is little information to what extent exome sequencing is a real substitute for a gene panel or for Sanger sequencing. On a larger scale, this issue is an important point in the field of diagnostic exome sequencing. What are the standards that the exome data are held to? Are we happy to consider exome sequencing a trial and error technology that is celebrated when successful but not mentioned when no mutation was found? I am looking forward to the first studies that provide a larger overview of the actual success rates of exome sequencing in recessive families, as the unsolved cases are usually not reported. Therefore, the authors do not assess the utility of exome sequencing, but merely provide an example that this method may find the culprit gene with relatively little effort.
Lessons learned. The successful gene identification in the publication by McDonell and collaborators is probably due to a combination of factors. Data analysis is relatively straight forward in (1) familial cases with (2) recessive inheritance and (3) a well-defined phenotype with (4) a known list of candidate genes. In the field of epilepsy, we rarely have such an ideal situation, as the number of possible candidate genes is larger and recessive epilepsies outside neurometabolic, storage, or neurodegenerative disorders are rare. Therefore, we will invariably end up with a longer list of variants. Basically, unless recessive inheritance is assumed and unless the phenotypes guide us to a list of candidates, the single exome approach might be misleading and not sufficiently informative.
Child Neurology Fellow and epilepsy genetics researcher at the Children’s Hospital of Philadelphia (CHOP), USA and Department of Neuropediatrics, Kiel, Germany


Pingback: Imbalance of a rare second messenger – FIG4 mutations in polymicrogyria | Beyond the Ion Channel
Pingback: SLC25A22, migrating seizures and mitochrondial glutamate deficiency | Beyond the Ion Channel
Pingback: The OMIM epileptic encephalopathy genes – a 2014 review | Beyond the Ion Channel