
MPP. Mitochondria are indispensable for cellular energy production and require constant protein import, as most mitochondrial genes are encoded in the nucleus. In order for proper targeting, mitochondrial proteins have a specific presequence, which is removed once a protein has found its way into the mitochondria. This function is accomplished by the mitochondrial processing peptidase MPP, which is encoded by the PMPCA and PMPCB genes. In a recent publication in the American Journal of Human Genetics, we identified PMPCB as a novel gene for a complex neurodegenerative condition in childhood and discovered a new disease mechanism for neurological disorders. However, epileptic encephalopathy that initially led to the inclusion of our initial RES study was only one extreme of an unusual disease spectrum.
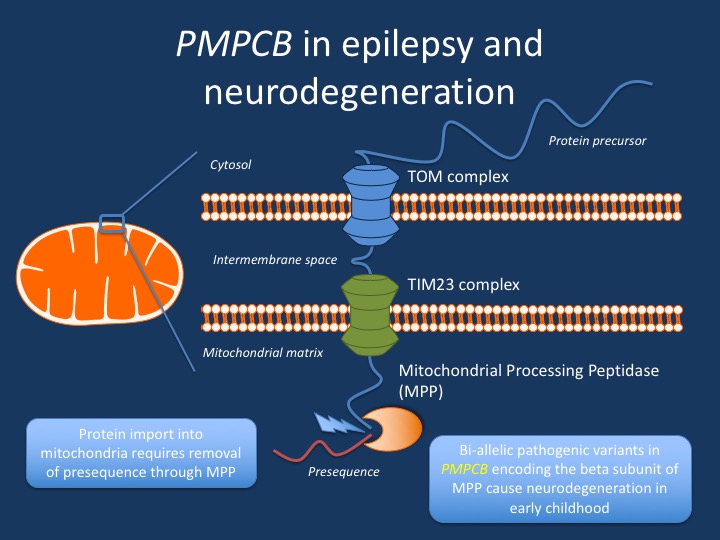
Figure. PMPCB dysfunction in childhood neurodegeneration and epilepsy results in reduced function of the mitochondrial preprocessing peptidase MPP. This protease cuts off the presequence that guides mitochondrial proteins to the mitochondrial matrix – MPP is the mitochondrial box cutter that opens up the packages addressed to the mitochondrial matrix.
Mitochondrial disease. In clinical practice, we typically assume that we do not encounter mitochondrial disease too often in patients with epileptic encephalopathies. We primarily assume ion channel diseases or disorders of synaptic transmission as main disease mechanisms. However, a subset of mitochondrial diseases can present with primary CNS presentation, defying the general assumption that mitochondrial diseases present with multisystem disease and clear biomarkers such as elevated lactate. Therefore, particularly in patients with regression or worsening during febrile illnesses or crises, mitochondrial diseases are always a differential diagnosis. This blog post is about the discovery of an unusual mitochondrial disorder that presented with epileptic encephalopathy in one of the initial EuroEPINOMICS families, which eventually led us to recognize a wider range of neurodegenerative conditions with prominent cerebellar atrophy.
The mechanism of PMPCB. When we first considered PMPCB as a candidate gene in a family from the EuroEPINOMICS project, we felt ambivalent about this gene as the cause of the disease. Little was known about this gene at the time, and the suspicious variant was a homozygous missense variant in a consanguineous Kurdish-Iraqi family – we typically encounter this class of variants on a regular basis and do not pay too much attention to them, as we typically see a large number of these variants in consanguineous families. On the other hand, PMPCB was a relatively intolerant gene – we had never seen variants in this gene before, which made us suspicious. When we connected with the Voegtle lab in Freiburg, Germany, it dawned on us why PMPCB variants occur so rarely – it encodes one subunit of MPP, the mitochondrial preprocessing protease, which is so critical to cellular functioning that it tolerates little variation. Basically, MPP is a mitochondrial box cutter that opens up the packages addressed to the mitochondrial matrix by removing a presequence. Yeast is usually a very robust single cell organism that tolerates a lot of genetic and environmental stress. Therefore, we were extremely surprised that the variant in the EuroEPINOMICS family basically took away the yeast’s ability to grow even under the most favorable conditions. When we examined patient cells from the affected individuals in the EuroEPINOMICS, we realized that mutated MPP is unable to fully process one of the most studied targets, the Frataxin protein that is involved in Friedreich’s ataxia. Mitochondria are no longer able to fully activate all the proteins addressed to them and unopened packages and letters keep accumulating. The mitochondrial box cutter has a very limited margin of error.
The breakthrough. The next step in the discovery of PMPCB occurred when we started collaborating with the Van Hove lab in Colorado, who had identified a patient with compound heterozygous PMPCB variants. Interestingly, this patient had a different phenotype with prominent cerebellar neurodegeneration. However, this patient did not have the epileptic encephalopathy that was initially seen in the EuroEPINOMICS family – epilepsy was in fact only the tip of the iceberg that made us realize a much wider range of phenotypes afterwards. We then soon identified two other patients with similar neurodegenerative phenotypes and variants that overlapped between patients. Biochemical studies showed mitochondrial dysfunction, including elevated lactate and impairment of the respiratory chain complexes. In addition, these variants did not lead to a lethal yeast phenotype and could finally be modelled.
The PMPCB yeast phenotype. It took me a while to understand how precisely PMPCB dysfunction can be analyzed in yeast. Yeast can be grown at low and high temperatures and in yeast strains carrying patient mutations, the PMPCB phenotype only becomes evident when yeast are stressed at higher temperatures. This situation mimics the increased energy demand with infections or with growth, possibly explaining why PMPCB-related conditions only manifest with infections or increased cellular demands during infancy. In addition, we were able to assess how MPP dysfunction affects various cellular functions. Interestingly, we found a very early and surprisingly strong impairment of iron-sulfur cluster biosynthesis. Iron-sulfur proteins are involved in a variety of cellular processes, most importantly the electron transport chain. PMPCB dysfunction is so dramatic that it impacts the generation of these proteins almost immediately once yeast cells are stressed with higher temperatures.
MPP diseases. Identifying the role of PMPCB dysfunction almost took a decade and during this time, mutations in PMPCA have been discovered in patients with cerebellar ataxia. PMPCA encodes the non-catalytic subunit of MPP that is involved in recognizing the protein needs to be cleaved – PMPCA is the handle of the mitochondrial letter opener while PMPCB is the blade. Taken together, both disorders affecting MPP function share a common theme – prominent cerebellar involvement in the absence of multisystem disease. The latter is surprising given the known presentations of other diseases that impair iron-sulfur cluster biosynthesis. Understanding why MPP dysfunction has such a strong effect on the cerebellum is not understood, but it may provide some insight on why other brain regions are somewhat protected against MPP dysfunction.
What you need to know. Bi-allelic variants in PMPCB are a novel cause of neurodegeneration in early childhood, and some patients can have prominent epileptic encephalopathy. While the impairment of mitochondrial function is due to an impairment of iron-sulfur cluster biosynthesis, patients with PMPCB-related disorders have not been found to have systemic symptoms. Prominent cerebellar atrophy in early childhood is one of the most specific features of PMPCB-related disorders.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
