
DEPDC5. We have selected DEPDC5 to be our gene of the week. DEPDC5 is currently the most common known gene for focal epilepsies. DEPDC5 mutations cause familial focal epilepsy with variable foci, an epilepsy syndrome with autosomal dominant inheritance where the affected family members can have different types of focal epilepsies, most frequently frontal lobe epilepsy. Despite seizure semiology that varies among family members, it is constant for each individual.
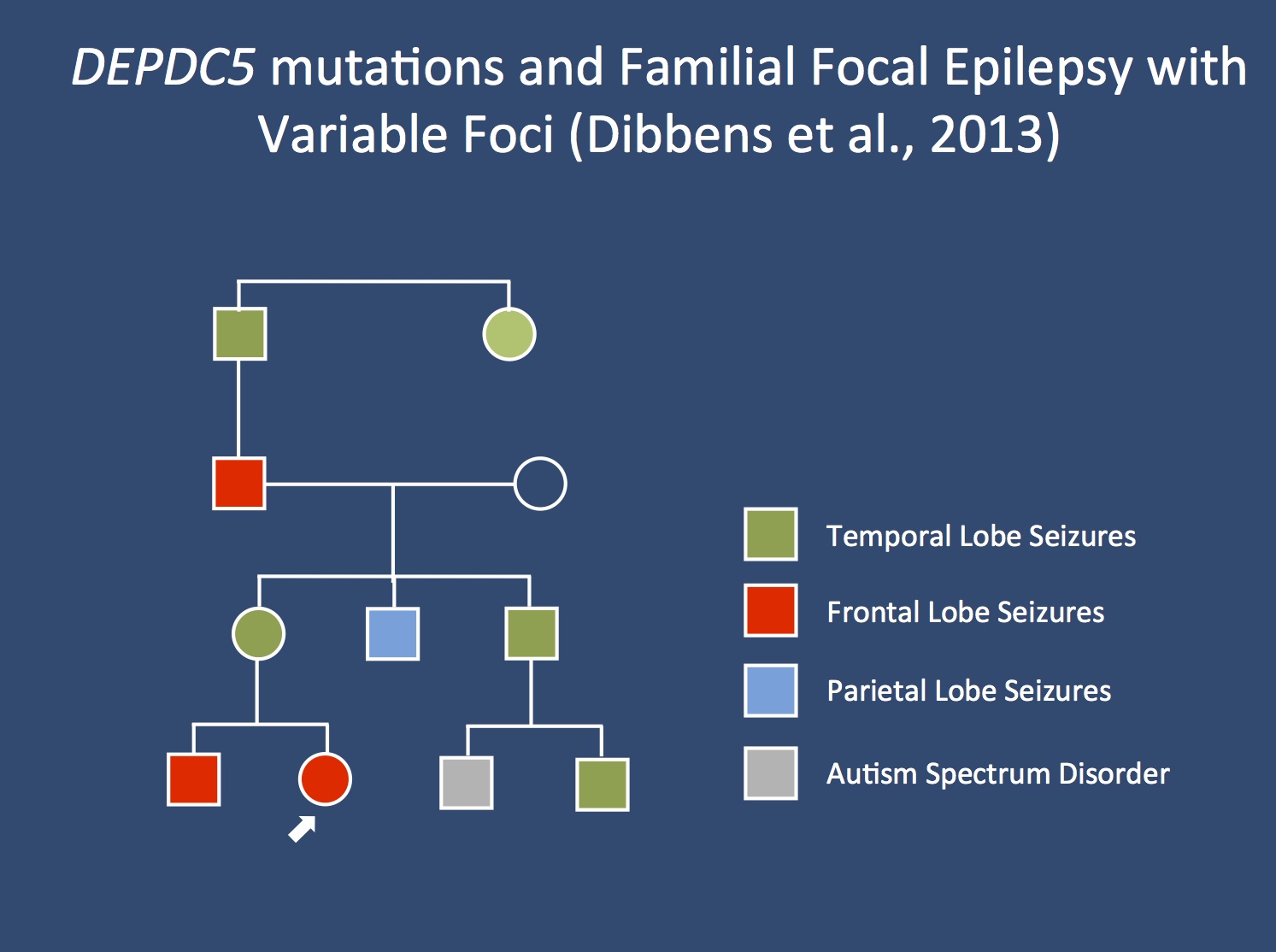
Pedigree A1 of the paper by Dibbens et al. In families with Familial Focal Epilepsy with Variable Foci, different focal epilepsies can be found in affected family members. In this family, affected individuals have temporal lobe seizures, frontal lobe seizures and parietal lobe seizures.
Familial focal epilepsy with variable foci. Mutations in DEPDC5 were first identified in familial focal epilepsy with variable foci. This epilepsy syndrome follows autosomal dominant inheritance and affected individuals have focal epilepsy. However, the location of the epileptogenic zone is variable in the different affected family members, e.g. some family members have frontal lobe epilepsy, others temporal lobe epilepsy etc. Usually, the location of the focus remains stable during life. Variability is present regarding the severity of the epilepsy and response to medication. Some family members may have intellectual disability or autism spectrum disorders. Structural imaging had long been considered to be normal, but recently focal cortical dysplasia has been found in some individuals with familial focal epilepsy with variable foci.
Autosomal dominant nocturnal frontal lobe epilepsy. This epilepsy syndrome is characterized by nocturnal seizures with frontal lobe semiology, e.g. hypermotor, tonic or dystonic features. The seizures typically arise from stage 2 sleep and frequently occur in clusters of multiple seizures per night. Differentiation from sleep disorders can be difficult in some cases and may require video EEG monitoring. Seizures during wakefulness are rare. Affected members of families with DEPDC5 mutations can by chance all have frontal lobe epilepsy mimicking the phenotype of autosomal dominant nocturnal frontal lobe epilepsy.
Familial mesial temporal lobe epilepsy. Familial mesial temporal lobe epilepsy is characterized by a mesiotemporal seizure semiology, e.g. psychic features, like for example déjà vu or epigastric aura (rising gastric sensation), in at least two family members with the same symptoms. However, DEPDC5 mutations are only rarely found in families where all affected individuals have mesial temporal lobe epilepsy.
Other syndromes. Furthermore, DEPDC5 mutations are seldom found in patients who have been diagnosed with childhood epilepsy with centrotemporal spikes, and may be absent in families with autosomal dominant lateral temporal epilepsy, a syndrome characterized by auditory or aphasic auras.
Genetics. Familial focal epilepsy with variable foci has been known as an epilepsy syndrome for more than a decade, but the causative gene remained a mystery for a long time. Linkage to chromosome 22q was established in 1999. The development of exome sequencing and the reevaluation of the original Australian family indicating linkage to the same chromosome 22q region, as in the other families, paved the way for the gene discovery in this and other families. DEPDC5 mutations were eventually identified in 7/8 large families with familial focal epilepsy with variable foci. Subsequently, mutations in DEPDC5 were also found in autosomal dominant nocturnal frontal lobe epilepsy and familial temporal lobe epilepsy, further supporting that DEPDC5 mutations can result in different focal epilepsies. As most mutations result in premature termination of the protein, haploinsufficiency is considered to be the disease mechanism. DEPDC5 mutations also account for 5–12% of smaller focal epilepsy families and may occur de novo in sporadic patients with focal epilepsy. The penetrance is 0.5-0.8 i.e. the likelihood that an individual with a DEPDC5 pathogenic mutation develops focal epilepsy is 50-80%.
Mechanism. The mTOR (mechanistic target of rapamycin) pathway regulates cell growth and cell proliferation. DEPDC5 is part of the GATOR1 subcomplex which modulates mTOR activity dependent on the availability of amino acids. Haploinsufficiency of DEPDC5 results in decreased inhibition of mTOR eventually leading to cortical dysplasia in variable locations. This mechanism is supported by the observation of cortical dysplasia in some individuals and families with DEPDC5 mutations. However, other individuals with DEPDC5 mutations have normal high-resolution imaging. It is currently unclear if these individuals also have cortical dysplasia which is too small to appear on MRI or if DEPDC5 mutations may also cause focal epilepsy by other mechanisms. It has been suggested that, similar to other mTORopathies, a “2-hit model” may be operative in DEPDC5 associated cortical dysplasia, assuming an additional somatic mutation in an mTOR pathway gene in addition to the DEPDC5 mutation; however, this hypothesis has not been confirmed. One group has recently identified families with mutations in two mTOR regulators: the NPRL2 and NPRL3 genes, suggesting that mutations in all three genes encoding proteins that make up the GATOR1 complex may influence focal epilepsy risk. Additional studies to confirm these observations will be required.
Therapy. Therapeutic approaches for patients with DEPDC5 mutations include all standard options, including drug treatment and potentially epilepsy surgery. It has been suggested that treatment with mTOR inhibitors such as rapamycin may also be helpful. mTOR inhibitors are used in tuberous sclerosis, another mTORopathy, but there are at present no studies supporting the use in patients with DEPDC5 mutations.