
Beyond de novo. One of the most robust ways to interpret exome data is the analysis of de novo mutations. However, in addition to the 1-2 de novo events that we can identify in every individual, there is a plethora of inherited variants that often look suspicious. Unfortunately, other than looking at monogenic recessive disorders, we are often incapable of understanding the importance of these inherited variants and tend to ignore them. A recent publication in Nature now overcomes this difficulty by applying a joint analysis of inherited and de novo variants in autism.
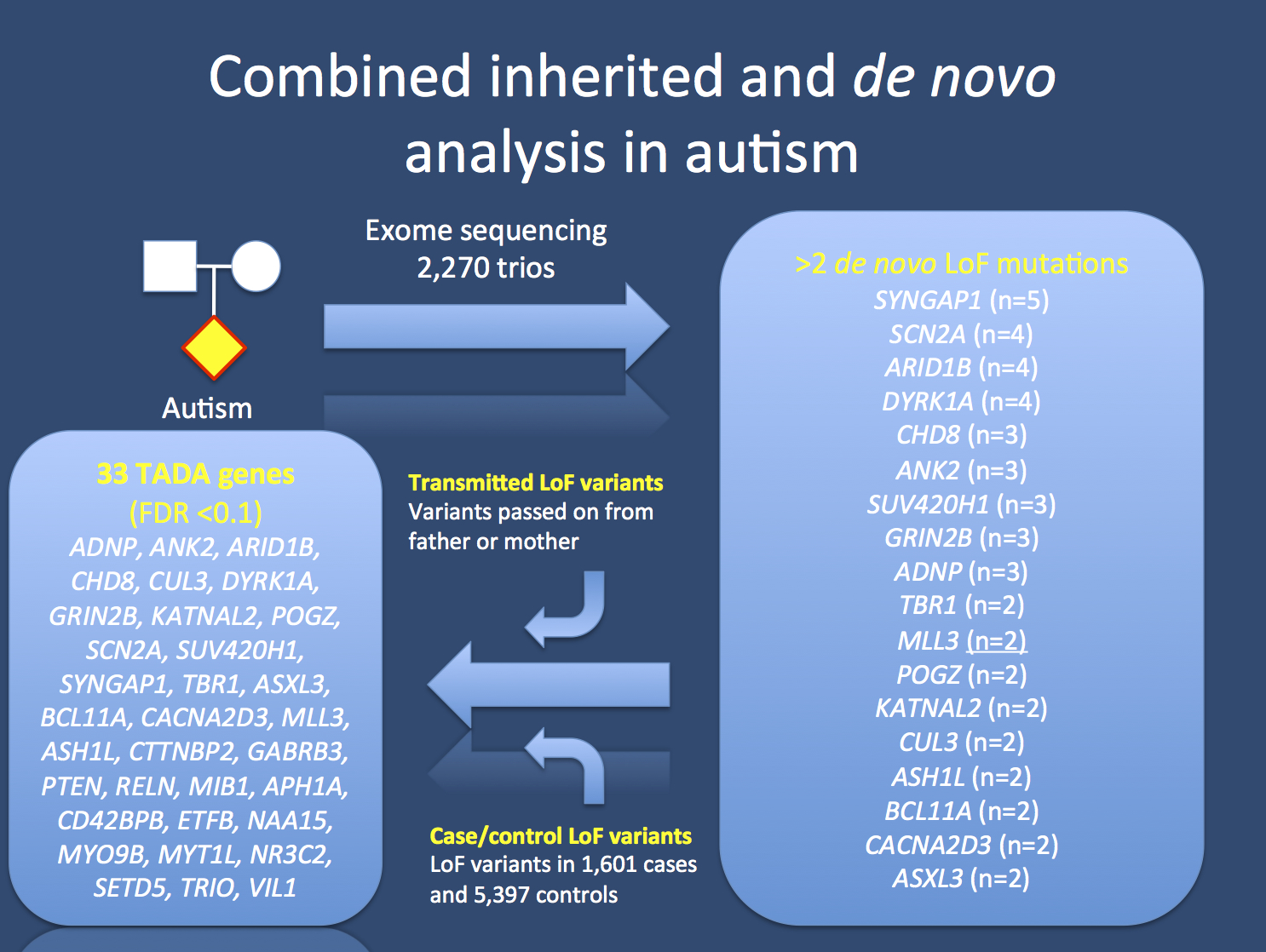
Concept of the recent Nature publication on autism. The gene list of 18 genes with two or more de novo loss-of-function mutations (LoF) is vastly expanded if the information from transmitted variants in patient-parent trios and information from case-control samples is added using the TADA framework. This results in a list of 33 genes or 107 genes, depending on the threshold used. The full list of 107 TADA genes can be seen here (link). FDR stands for false discovery rate and is a measure on the false positive rate in these types of tests.
Coincidence. There are several coincidences, which made me chose last November’s Nature publication on autism for this post. First, we just came out of a teleconference on one of our epilepsy genes yesterday where we discussed the role of inherited variants in epileptic encephalopathies. Secondly, we had found a damaging de novo mutation in one of this publication’s candidate genes in one of our Kiel patients late last year. Third, I finally picked up the publication yesterday that was lying on my desk for the last six weeks. Even though I had given it several attempts before, this was the first time that I felt that I understood what it was all about. Here is the story about TADA.
Limitations of de novo. The story of the publication of a large consortium of autism researchers starts with a sobering observation. The authors sequenced almost 2,500 patient-parents trios and only found 18 genes with more than one de novo loss-of-function (LoF) mutation. While this is more than expected by chance, it is not a very satisfying observation. First, using stats, approximately two of these genes might have simply occurred by chance. Secondly, the proportion of cases explained through these de novo variants is very small. As the authors also observed an excess of inherited variants in these candidate genes, it raised their suspicion that aiming for a combined analysis of de novo and inherited variants may help delineate candidate genes in a more detailed manner. This was also somewhat a necessity for them as most of the > 15,000 samples they had access to were not sequenced as patient-parent trios. Therefore, they looked for a statistical model that was able to include both types of variation.
TADA. The transmission and de novo association test (TADA) provided the authors with a framework to include de novo, transmitted, and case-control variation in their samples. It is a test that weighs certain types of variation in the analysis, and a de novo loss-of-function mutation was weighted more heavily than a de novo missense mutation, which in turn was more heavily weighted than a transmitted loss-of-function mutation. In a previous methods paper, it was already shown that this method is more powerful to detect candidate genes than the analysis of de novo mutations alone. In brief, using this model, the list of candidate genes was both expanded and stratified with respect to statistical certainty. Depending on the cut-off the authors used, they identified a list of 33 or 107 genes, referred to as the TADA genes (link). And this is where the story really gets interesting.
107 candidates. The TADA analysis basically expands the list of genes in which de novo mutations were found and refines this list for candidate genes. Interestingly, many known genes for neurodevelopmental disorders are included in this list, which would not have been detected otherwise. For example, SHANK3 suddenly becomes significant, even though there was only a single case with a LoF de novo mutation. The supporting evidence for SHANK3 stems from two transmitted LoF variants and an excess of LoF variants in cases (0.06%) compared to controls (0%). The same applies to genes such as RELN and NRXN1. In other established genes such as TRIO, the transmitted LoF variants compensate for a small increase of LoF variants in cases compared to controls. And there are also genes in which only the case-control comparison adds to the overall significance, as in the case of GABRB3, which initially didn’t rise above genomic noise with one loss-of-function and one missense mutation. GABRB3 is significant due to a prominent overrepresentation of loss-of-function variants in cases (0.12%) compared to controls (0.2%). The authors perform various pathway analyses on the TADA genes and establish that these genes basically fall into three big categories: synaptic transmission, chromatin remodeling, and transcriptional regulation.
What you need to know about TADA. We have ignored transmitted variants so far unless they point towards a monogenic recessive disease. The basic concept of the combined de novo and transmission analysis is the realization that candidate genes for neurodevelopmental disorders may confer different types of risk to disease. Some variant may be causative (de novo), others only contributory and transmitted. Including the information on the inherited variants may therefore help find causative genes in situations when a single de novo mutation is insufficient. This framework can easily be applied to other data sets such as our combined E2 epilepsy data set or the joint EuroEPINOMICS-RES data set, combining this with case control data that we have accumulated in other epilepsies such non-lesional focal epilepsies and IGE/GGE. I would even go as far as saying that this type of systematic joint analysis will be one of our major tasks in the future.
Child Neurology Fellow and epilepsy genetics researcher at the Children’s Hospital of Philadelphia (CHOP), USA and Department of Neuropediatrics, Kiel, Germany

