
SYNGAP1. This is the Epilepsiome page of SYNGAP1, a gene for intellectual disability, autism, and generalized epilepsy. SYNGAP1 was initially discovered in 2009 and has become a prominent gene for these conditions.
Here are the most recent blog posts on SYNGAP1
- Balanced translocations in neurodevelopmental disorders
- Less is more – gene identification in epileptic encephalopathies through targeted resequencing
- Typical versus atypical: exome sequencing in pediatric epilepsies
- The 1003 possible autism genes – a matter of constraint
In a nutshell. SYNGAP1 encodes a regulatory protein of the postsynaptic density that is involved in neuronal plasticity. De novo pathogenic variants in SYNGAP1 have been observed in patients with non-syndromic intellectual disability, autism, and patients with epileptic encephalopathy, mainly with generalized seizure types.
| Phenotypes | Genetics | Mechanism & Function |
| The Clinical Perspective | Community | Resources & References |
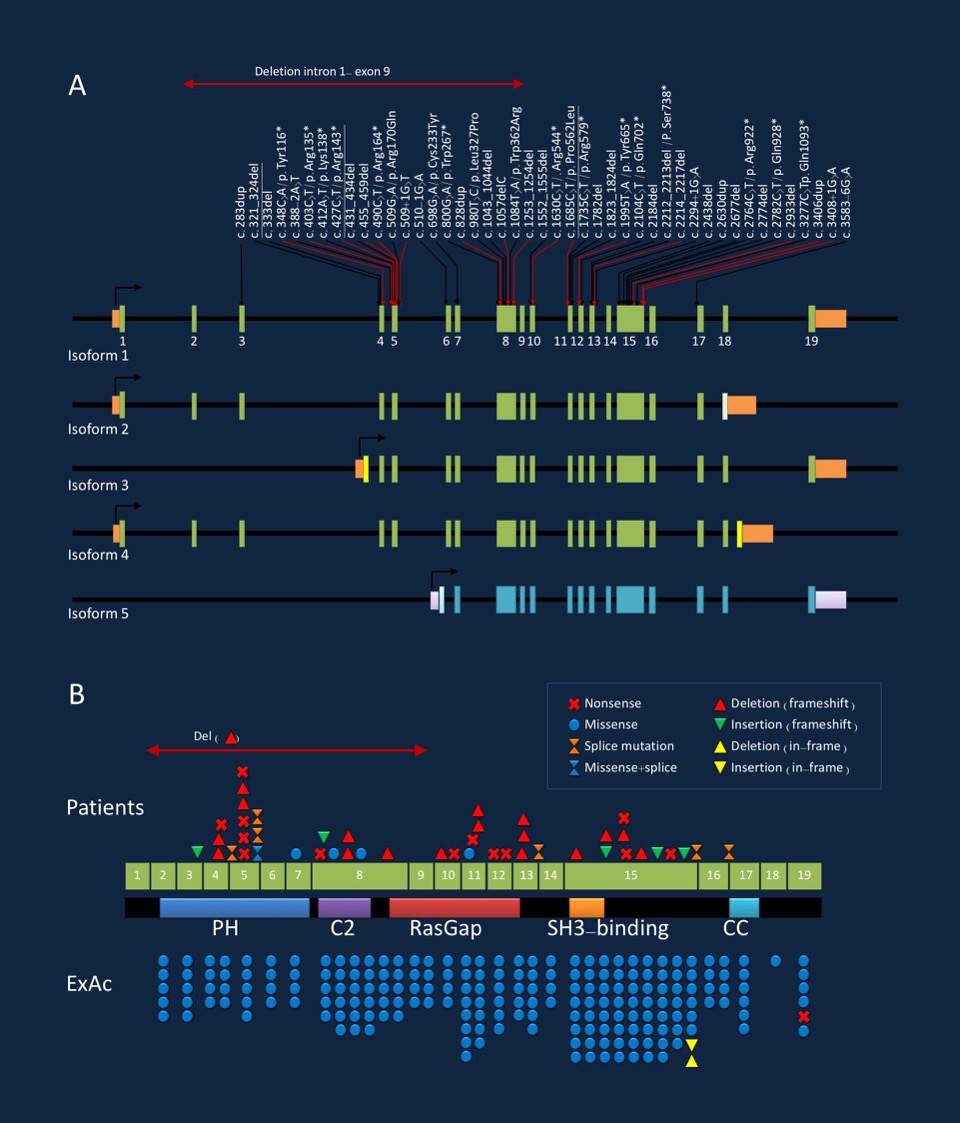
Figure 1. Pathogenic variants in the SYNGAP1 gene with respect to isoforms (A) and protein structure (B). Figure refers to publication by Mignot and collaborators. Figure modified from draft figure provided by the author.
Phenotypes
Phenotypes. SYNGAP1 is primarily associated with three main phenotypes, including non-syndromic intellectual disability, autism, and epileptic encephalopathy. Many patients have a combination of these features.
Intellectual disability and autism. The full spectrum of the severity of intellectual disability associated with SYNGAP1 is not yet fully characterized. In our current study by Mignot and collaborators, we found that more than half of all patients have severe intellectual disability, while the other half of patients have milder degrees of intellectual disability. A diagnosis of autism spectrum disorder is present in roughly 50% of patients.
Epilepsy. In the recent study by Mignot and collaborators, seizures appear to be a very common feature in patients with pathogenic SYNGAP1 variants. Patients showed generalized seizures types including atonic or myoclonic seizures, absences and myoclonic absences and eyelid myoclonia. A significant fraction of patients were photosensitive. These seizures types are not always easily classified into existing epilepsy syndromes, but sometimes patients are diagnosed with a myoclonic astatic epilepsy (MAE, Doose Syndrome) or Epilepsy with Myoclonic Absences (EMA). The EEG frequently shows generalized activity. Seizure severity does not seem to correlate with autism or the degree of cognitive impairment. Hypotonia and unstable gait are also common features.
Genetics
Mutation spectrum. Truncating and missense variants, as well as deletions, have been reported in patients with SYNGAP1 encephalopathy. There are some recurrent variants in the SYNGAP1 gene including the variants c.321_324del, c.427C>T/p.Arg143*, and c.1685C>T/p.Pro562Leu. Pathogenic variants in the SYNGAP1 gene are located throughout the gene, although some exons are spared. SYNGAP1 is a gene with a very large number of different isoforms. This is taken as an explanation why some exons in the SYNGAP1 appear to be relatively spared from mutations.
Genotype-phenotype correlation. There is no known genotype-phenotype correlation in the SYNGAP1 gene. It will be interesting to see in the future whether patients with recurrent pathogenic variants have comparable phenotypes. In many genetic epilepsies, even patients with identical variants can have a wide range of associated clinical features. In the study by Mignot and collaborators, there was some indication that patients with pathogenic variants in exons 4-5 are pharmacosensitive, while patients with variants in exons 8-15 tend to have intractable seizures.
General Considerations for Variant interpretation. When reviewing a genetic variant to determine its significance for a given patient, it is important to weigh multiple pieces of evidence:
Considerations for gene level interpretation. First, it is important to establish the strength of the evidence showing that the gene is associated with epilepsy. Some genes may only have one variant reported in a single individual with epilepsy, while other genes may have multiple variants reported in many large families with an autosomal dominant pattern of epilepsy. For SYNGAP1, there is strong evidence for its role in human epilepsy.
Considerations for variant level interpretation. When reviewing the significance of a variant, it is important to consider the impact on the gene and the presence of the variant in previously described patient and control populations. Many clinical genetic testing laboratories classify genetic variants into different categories, ranging from benign to pathogenic. Variants that are common in control populations and would not be predicted to have a major impact on the gene/protein are generally classified as benign. Variants are more likely to be classified as pathogenic if the variants are rare or not present in the control population, reported in multiple individuals or families with disease, and likely to have a higher impact on the gene/protein based on the type of mutation or functional studies. Variants with uncertain or limited available evidence may be classified as variants of uncertain significance (VUS), indicating that further information is required in order for the variant to be further defined. In some cases, testing additional family members can be helpful, as it allows the lab to determine whether or not the variant was inherited (versus de novo) and how the variant segregates with disease in the family. This is particularly true for SYNGAP1 variants, as most disease-causing variants would be expected to be de novo. Sometimes further classification of a VUS requires waiting for the identification of additional patients or families with similar or nearby variants.
Inheritance, Penetrance & Prevalence. Most pathogenic variants in the SYNGAP1 gene are de novo. Considerable variable expressivity can occur, even with the same variant, particularly in the severity of intellectual disability and the presence of seizures and autism. The exact prevalence of SYNGAP1-related conditions is unknown. According to one recent large study, SYNGAP1 variants may be identified in as many as 0.5-1% of children with neurodevelopmental disorders.
Mechanism & Function
Mechanism. SYNGAP1 encodes for an essential protein of the post-synaptic density (PSD). The SYNGAP1 protein is a regulatory component of the postsynaptic density that is involved in neuronal plasticity. The effect of SYNGAP1 protein on the maturation of dendritic spines appears to inhibitory and reduced or absent SYNGAP1 protein may lead to enhanced maturation of dendritic spines. The SYNGAP1 protein is strategically placed in the postsynapse to be an interface between the NMDA receptors at the postsynaptic membrane and the signaling apparatus in the postsynapse that modulate the architecture of the dendritic spine. The underlying mechanism in SYNGAP1 encephalopathy is haploinsufficiency, i.e. the lack of two functional copies of the SYNGAP1 gene.
Cell/Animal models.
Mice. Mouse models completely deficient for SYNGAP1 are non-viable. Mouse models with decreased expression of SYNGAP1 have behavioral and cognitive issues.
The Clinical Perspective
Recurrence risk & testing of family members. SYNGAP1 variants are generally de novo, although at least one individual has been reported to inherit a pathogenic SYNGAP1 variant from a parent with somatic mosaicism. Each child of an individual with a disease-causing variant has a 50% (1 in 2) of inheriting the disease-causing variant. Although not yet reported, germline mosaicism of SYNGAP1 variants is possible, making the recurrence risk for families with one child with a SYNGAP1 variant higher than the general population risk.
Therapy. There is no gene-specific treatment available for SYNGAP1 at this point. Given that patients usually present with seizures in late infancy (mean age of onset 12 months), patients are usually treated with antiepileptic drugs for generalized epilepsies including valproic acid, lamotrigine, levetiracetam. There are no specific antiepileptic medications that are known to exacerbate seizures.
Community
Research studies. The scientific community is actively studying SYNGAP1 and its role in human disease. The SYNGAP1 Epilepsiome team is happy to facilitate if you have questions or a specific interest in this gene.
Family connections. The SYNGAP1 community is very active and the Bridge the Gap-SYNGAP Education and Research Foundation maintains a website about the most recent findings and research activities regarding SYNGAP1-related conditions.
Patient registry. The Bridge the Gap-SYNGAP Education and Research Foundation has partnered with Simons VIP Connect and the Rare Epilepsy Network on their registries.
Resources & References
Websites for Families
SYNGAP1 on National Organization for Rare Disorders
Websites
References
Mignot C et al. 2016 Genetic and neurodevelopmental spectrum of SYNGAP1-associated intellectual disability and epilepsy. J Med Genet. 2016 Mar 17. pii: jmedgenet-2015-103451. doi: 10.1136/jmedgenet-2015-103451. [Epub ahead of print] PMID: 26989088
Berryer MH et al. 2013 Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum Mutat. 34(2):385-94. PMID: 23161826
Hamdan FF et al. 2009 Mutations in SYNGAP1 in autosomal nonsyndromic mental retardation. N Engl J Med. 360(6):599-605. doi: 10.1056/NEJMoa0805392. Erratum in: N Engl J Med. 2009 Oct 29;361(18):1814. Perreault-Linck, Elizabeth [corrected to Perreau-Linck, Elizabeth]. PMID: 19196676