
Shouldering the Load. The CLTC gene encodes the protein clathrin, which plays a crucial role in the formation of clathrin-coated vesicles, responsible for transporting proteins and other molecules within neurons. Clathrin-mediated endocytosis is also a crucial process for the recycling of synaptic vesicles in neurons, enabling efficient neurotransmitter release and synaptic transmission. The discovery of CLTC-related disorders has revealed a diverse spectrum of neurological conditions, ranging from intellectual disability to epilepsy. Here is a blog post on CLTC-related disorders as the forgotten disease of synaptic vesicle recycling, highlighting the crucial role of clathrin in maintaining proper neurological function.
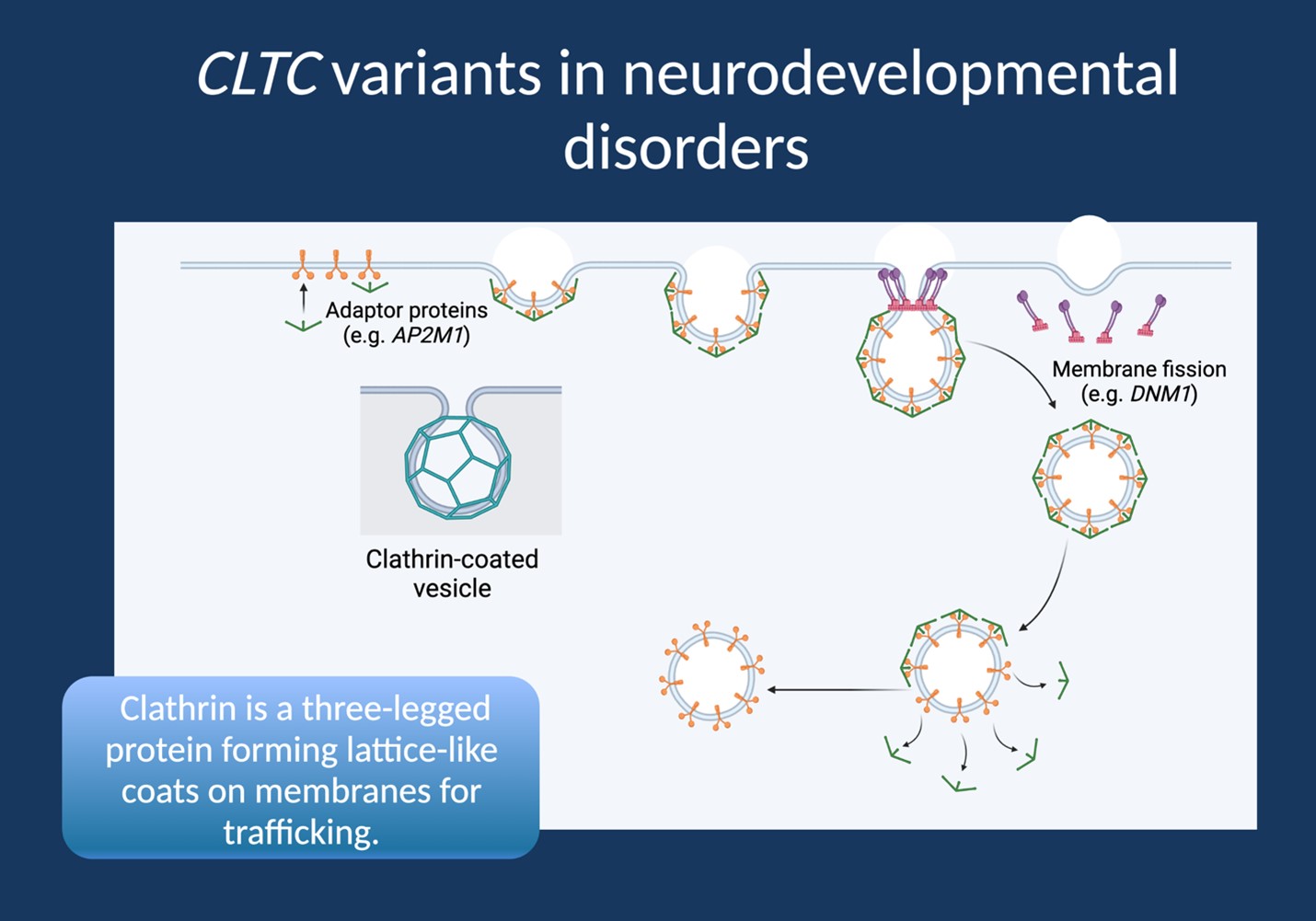
Figure 1. Clathrin-mediated endocytosis is a fundamental process for the recycling of synaptic vesicles in neurons, enabling the efficient release of neurotransmitters and maintaining proper synaptic function. The process involves the formation of clathrin-coated pits, which are then internalized and recycled to form new synaptic vesicles. Clathrin interacts with a host of other proteins to form a complex machinery that drives this process. Mutations in genes encoding these proteins, including CLTC, can disrupt the formation and function of clathrin-coated pits and impair synaptic vesicle recycling. This disruption can lead to various neurological disorders, such as epilepsy and intellectual disability.
Decoding the CLTC mystery. The discovery of clathrin and its role in synaptic vesicle recycling has revolutionized our understanding of synaptic function. In addition to clathrin, other proteins, including AP2M1 and DNM1, have been implicated in the regulation of clathrin-mediated endocytosis. AP2M1 is a component of the AP2 complex, which binds to clathrin and recruits it to the plasma membrane. DNM1, on the other hand, is a GTPase that functions in the fission of clathrin-coated vesicles from the plasma membrane. Mutations in the genes encoding these proteins have been associated with various neurological disorders, further underscoring the critical role of clathrin-mediated endocytosis in synaptic function. Importantly, the study of these proteins has shed light on the complexity of the synaptic vesicle recycling process, which involves a tightly regulated interplay between various proteins and signaling pathways.
CLTC-related disorders. In recent years, several studies have identified a link between CLTC mutations and various neurodevelopmental disorders. One of the earliest studies, conducted by Michael J. Friez et al. and published in the American Journal of Human Genetics in 2016, reported a correlation between de novo mutations in the CLTC gene and intellectual disability, epileptic encephalopathy, and microcephaly. Subsequently, Homa Tajsharghi et al. published a study in the same journal in 2018, identifying a specific CLTC mutation associated with a syndromic form of developmental and epileptic encephalopathy. This finding provided further evidence of the role of clathrin in neurological disorders. Finally, a study by Avinash Abhyankar et al. published in Science Translational Medicine in 2019, identified additional CLTC mutations linked to neurodevelopmental and neurodegenerative disorders, including intellectual disability and motor impairment. These studies highlight the growing recognition of CLTC-related disorders and the need for further research to understand the underlying mechanisms of these conditions.
Clinical features. CLTC-related disorders are characterized by a range of clinical features, including intellectual disability, developmental delay, epileptic encephalopathy, microcephaly, and motor impairment. Additionally, affected individuals may exhibit hypotonia, ataxia, spasticity, and behavioral problems such as autism spectrum disorder. While the exact number of individuals with CLTC-related disorders is unknown, several cases have been reported in the literature. These studies suggest that CLTC-related disorders are a rare but significant cause of neurological dysfunction, with a range of clinical presentations and varying degrees of severity. Further research is needed to better understand the molecular mechanisms underlying these disorders and to develop targeted therapies for affected individuals.
Difference from other synapse disorders. While both CLTC-related disorders and other synapse disorders can lead to neurological dysfunction, the clinical features and underlying mechanisms may differ. For example, some synapse disorders are associated with abnormal synaptic plasticity, while CLTC-related disorders are associated with defects in synaptic vesicle recycling. Additionally, the clinical presentation of CLTC-related disorders may be distinct from other synapse disorders, with features such as microcephaly and motor impairment being more common in CLTC-related disorders. For example, CLTC-related disorders and STXBP1-related disorders are both neurodevelopmental disorders that can cause intellectual disability, epilepsy, and other neurological symptoms. However, STXBP1-related disorders tend to be more severe than CLTC-related disorders. This is because STXBP1 is a key regulator of neurotransmitter release, and mutations in the STXBP1 gene can cause defects in synaptic transmission that affect many different types of synapses throughout the brain. In contrast, CLTC-related disorders are primarily associated with defects in synaptic vesicle recycling, which affects a more limited set of synapses. Further, while both types of disorders can cause seizures, STXBP1-related disorders tend to be associated with more severe and refractory seizures that are difficult to control with medication. Finally, the severity of a neurodevelopmental disorder can also depend on the specific mutation and its effect on protein function.
The forgotten synapse gene. In our ENGIN teaching sessions, we often refer to CLTC-related disorders as one of the “forgotten disorders” of synaptic vesicle recycling because they are relatively less studied and less well-known compared to other synaptic vesicle disorders, such as those related to STXBP1 or SYNGAP1. This may be due in part to the fact that CLTC mutations are relatively rare and were only recently identified as a cause of neurodevelopmental disorders. Additionally, the symptoms of CLTC-related disorders can be highly variable and may overlap with other neurodevelopmental disorders, making diagnosis and treatment challenging. Finally, the role of clathrin in neuronal function is complex and not fully understood, further contributing to the challenge of studying CLTC-related disorders. Nevertheless, continued research into the genetic and molecular mechanisms underlying CLTC-related disorders is essential for improving our understanding of synaptic vesicle recycling and developing targeted therapies for affected individuals.
Written jointly by Sarah Ruggiero and Ingo Helbig
Sarah Ruggiero is a Genetic Counselor at the Epilepsy Neurogenetics Initiative and Helbig Lab, Children’s Hospital of Philadelphia.
