
PME. The progressive myoclonus epilepsies (PME) are an important and distinct subgroup of genetic epilepsies. In contrast to many genetic epilepsies with a neurodevelopmental trajectory, the PMEs often follow a neurodegenerative course, which is characterized by a worsening myoclonus over time and frequently associated with cognitive decline. In a recent publication, protein-truncating variants in the intronless gene IRF2BPL were identified in two individuals with PME. However, in contrast to the relatively distinct nature of most other PME, the clinical presentation in IRF2BPL-related disorders is part of a phenotypic spectrum and emerges as one of the most usual phenotypic discoveries in the genetic epilepsies to date.
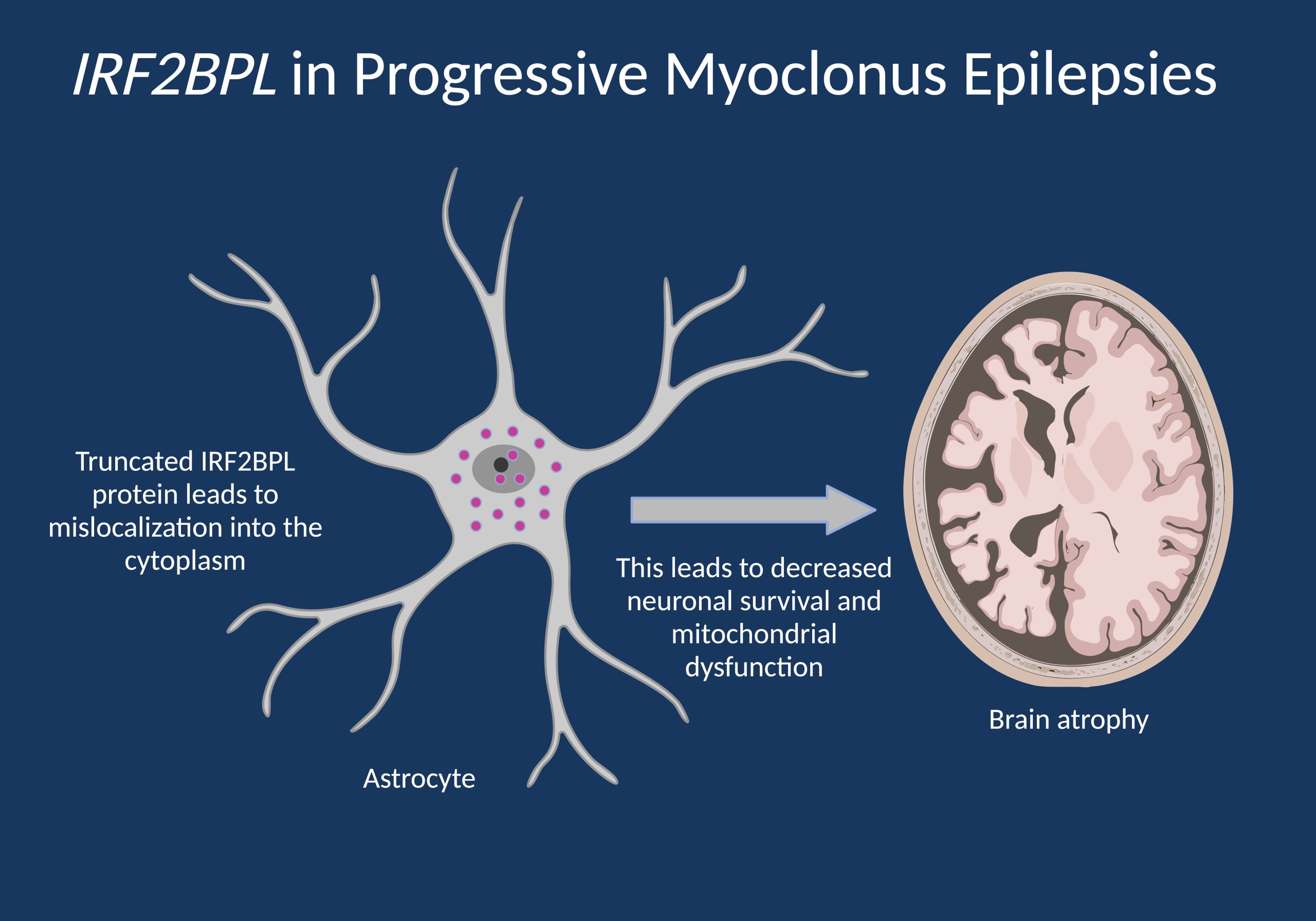
Figure 1. IRF2BPL is an intronless gene, and protein-truncating variants do not result in nonsense-mediated decay (NMD) but atypical dimerization that leads to displacement of IRF2BPL from the nucleus to the cytoplasm. Given the likely role of IRF2BPL in transcriptional regulation, this results in decreased neuronal survival and mitochondrial dysfunction, explaining the progressive nature of IRF2BPL-related disorders.
The PMEs. Prior to discussing IRF2BPL-related disorders, it is worthwhile to review what is known about the group of conditions are referred to as the progressive myoclonus epilepsies (PME). The PMEs are a group of seizure disorders that initially present with therapy-resistant myoclonus that gets worse over time and can be accompanied by generalized tonic-clonic seizures and cognitive decline. The PMEs are usually thought of as a distinct group of epilepsies with a genetic basis and is usually divided into two larger groups. Individuals with Unverricht-Lundborg disease (ULD) typically have preserved cognition until later in the disease course, while individuals with disorders such as Lafora body disease show prominent neurological decline and dementia. Only a few years ago, the genetics of ULD-type PME was simple, as an expansion of a dodecamer repeat in the 5’-UTR of the Cystatin B (CSTB) gene was the only known genetic cause. However, with the discovery of SCARB2, PRICKLE1, GOSR2, and KCNC1, additional genes were found in ULD-type PMEs. The Lafora-type PMEs were traditionally thought of as more heterogeneous as several storage disorders including sialidosis, mitochondrial disorders, or neuronal ceroid lipofuscinosis may present as progressive myoclonus epilepsies.
IRF2BPL. In the recent publication by Costa and collaborators, protein-truncating variants were identified in two individuals with PME that started in adolescence after an initial period of typical development. Patient 1 presented with neurological symptoms starting at the age of 10 years that developed into a prominent action myoclonus. Patient 2 started having tremor and myoclonus at the age of 15 years. Symptoms in both individuals were progressive. In addition, in both individuals, no symptoms were present prior to the onset in adolescence. While such a description is relatively typical for individuals with PME, the finding by Costa and collaborators is surprising given what is already known about IRF2BPL-related disorders. In 2018, IRF2BPL-related disorders were first described by Marcogliese and collaborators in a complex neurological phenotype including regression, hypotonia, progressive ataxia, and epilepsy, but also in a milder phenotype. Subsequently, IRF2BPL protein-truncating variants were identified in movement disorders and developmental and epileptic encephalopathies with a variable onset between the first month of life and adulthood. Antonelli and collaborators recently grouped the phenotypes into three groups, including early-onset developmental and epileptic encephalopathies, childhood-onset movement disorders, and late-onset neurodegenerative features. What was not clear to date was that some individuals with IRF2BPL-related disorders have a progressive myoclonus epilepsy.
The phenotypic spectrum. Intrigued by the PME phenotype in their two index patients, the authors reviewed the existing literature on IRF2BPL-related disorders. They identified 13 additional individuals with myoclonic seizures. However, in most individuals, the clinical presentation started in early childhood, with regression and developmental delay as the initial manifestation and followed by myoclonus afterwards. Contrasting these presentations with the typical PME phenotypes in their patient cohorts, IRF2BPL-related disorders appear to have a very unusual phenotypic spectrum with an onset of degenerative features between the first year of life and adulthood. The authors did not identify any clear genotype-phenotype correlations and this spectrum of phenotypes is both surprising and unprecedented. Accordingly, IRF2BPL-related disorders require particular attention in diagnosis and clinical care, given that the phenotypic range and disease dynamic require further studies. This is relevant as such considerations will impact treatment strategies in affected individuals.
This is what you need to know. IRF2BPL-related disorders comprise a range of progressive neurological disorders with a highly variable age of onset and phenotypic features. The progressive myoclonus epilepsies are part of the IRF2BPL spectrum, and this gene is the only known disease gene to date that includes the PMEs as part of such a wide phenotypic spectrum. Given the unusual presentation of IRF2BPL-related disorders, this gene may be an important candidate to consider in individuals with unsolved progressive myoclonus epilepsies and childhood- or adolescent-onset neurodegeneration more broadly. Full knowledge on this wide phenotypic range will help in understanding how novel treatments can be used most optimally.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
