
MCDs. Malformation of cortical developments are a frequent cause of intractable epilepsies and, if appropriate, surgical resection may be warranted. Malformations represent a wide range of cortical lesions resulting from derangements of normal intrauterine developmental processes affecting the formation of the cortical mantle. Polymicrogyria (PMG) is one of the most common malformations of cortical development. However, while somatic mutations affecting the mTOR pathway are a known cause of certain subtypes of MCD, the polymicrogyrias have remained elusive. The underlying cause remains unknown in more than 80% of cases and, if identified, may be due to a wide range of underlying genetic causes. In a recent publication, mosaic trisomy 1q was identified as a novel and relatively frequent cause of polymicrogyria, emphasizing the role of somatic mutation detection in malformation of cortical development.
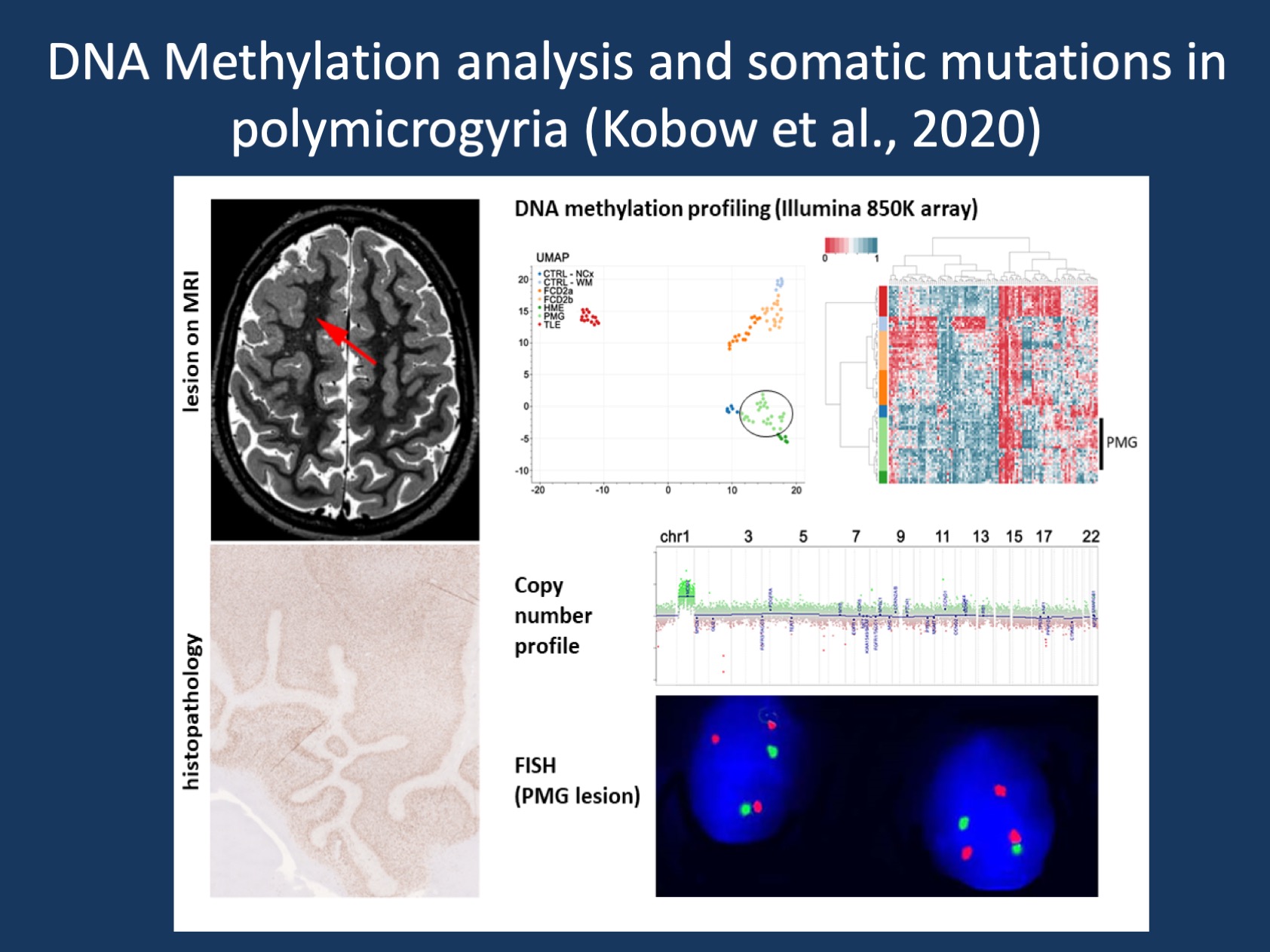
Figure 1. Brain mosaicism as new defining feature in a subset of PMG. Performing copy number profiling from DNA methylation data, Kobow and colleagues identified a somatic duplication of the entire long arm of chromosome 1 in surgical brain tissue of a subset of PMG patients. Subsequent FISH analysis identified a mosaic distribution of 1q triploid nuclei in the center of the lesion, but not in adjacent anatomically normal appearing cortical tissue (Figure provided by the authors).
Malformations of cortical development. While we often refer to “brain malformations” as a somewhat homogeneous group, the MCDs are extremely heterogeneous and the result of a wide range of pathological mechanisms. Generally, pathological features depend mainly on the timing of the defect in the developmental processes that may include abnormal proliferation or apoptosis, differentiation, neuronal migration, or layering. However, classification of MCD has proven challenging in everyday clinical practice. A definitive diagnosis of MCD is ideally based on neuropathological findings. Unfortunately, pathological tissue is not always available, and even if there is access to the lesion, inter- and intra-observer ratability may vary significantly among neuropathologists. To address this issue, either objectification of the histopathological workup is desired, e.g., using artificial intelligence and pattern recognition, or use of molecular diagnostic biomarkers.
PMG. Polymicrogyria is an MCD characterized by an excessive number of abnormally small, partly fused, and so called frustrane gyration, together with abnormal cortical lamination. Clinical signs and symptoms are variable depending on how many and which brain regions are affected. They may range from mild intellectual disability, mobility and language problems to severe encephalopathy with intractable epilepsy. A recently published international consensus recommendation on the diagnostic work-up for MCD recognized PMG as one of the most frequent and most heterogeneous cortical malformations in etiology with currently 9-20% of cases yielding a (blood-based) molecular diagnosis. On our blog, we have featured some of the rare genetic causes of PMG, such as the recurrent SCN3A p.I875T de novo variant. Tubulinopathies due to disease-causing variant in TUBA1A, TUBB2B, TUBB3, TUBB5 or TUBG1 are also a recurrent cause of PMG.
Molecular classification of structural brain lesions. There is growing evidence that integrating molecular signatures with histopathological phenotypes can be very helpful in the classification of structural brain lesions. This integration of molecular and histopathological features applies to many different brain tumors and also MCDs. Beyond disease-related genetic variants, DNA methylation classifiers have recently been identified as a valuable source in the decision-making process for disease diagnosis, prognosis, and treatment. DNA methylation is an epigenetic mark with a key function in developmental gene regulation, but also in phenotypic variability following environmental cues. Unsurprisingly, DNA methylation plays an essential role in brain development, function and disease. In the past, Kobow and colleagues had shown that genomic DNA methylation in the epileptic brain was highly different from that of healthy control tissue. Furthermore, DNA methylation signatures were highly etiology-dependent when comparing different rodent models of epilepsy. This study highlighted the diagnostic value of such epigenetic markers in the classification of epilepsies, which was previously unknown. The same group also showed that genomic DNA methylation signatures distinguished major subtypes of human Focal Cortical Dysplasia (FCD) from individuals with Temporal Lobe Epilepsy (TLE) compared to controls. In their recent study, Kobow and collaborators extended this finding to a new patient cohort and identified distinct methylation classes that differentiate between FCD among other MCD, TLE, and autopsy controls (Fig. 1).
Brain mosaicism as new defining feature in a subset of PMG. Performing copy number profiling from DNA methylation data, Kobow and colleagues identified a somatic duplication of the entire long arm of chromosome 1 in surgical brain tissue of a subset of PMG patients. This finding was both new and unexpected and was found in 7 out of 26 PMG patients. This may indicate that one in four individuals with unilateral PMG carry such a duplication, which would be much more frequent than any other single gene causes in PMG. Subsequent FISH analysis identified a mosaic distribution of 1q triploid nuclei in the center of the lesion, but not in adjacent anatomically normal appearing cortical tissue. This suggest that the 1q triplody was specific for PMG and not merely an accidental finding.
The 1q triploidy phenotype. Chromosome 1 is the largest human chromosome, with 2,055 coding and 2,092 non-coding genes. Several 1q related microdeletion and duplication syndromes or translocations are known. The clinical features of most individuals with 1q related copy number variations included developmental delay and learning disabilities, slow growth and short stature, various congenital disabilities, specific facial features, as well as neoplasias. This manifold of diseases highlights the central role of genes on 1q in overall development, but also for brain structure and function. The newly described subgroup of patients with somatic 1q triploidy presented clinically with a unilateral isolated PMG, early-onset focal epilepsy and a severe combinatorial developmental delay. So far, the only recurrent finding in individuals with MCD were somatic mosaicism of PI3K-AKT-MTOR pathway-related genes in FCD type 2 and hemimegalencephaly (HME). Most recently, brain mosaic SLC35A2 mutations were reported in FCD type 1, mild malformation of cortical development (mMCD), and non-lesional focal epilepsy.
What you need to know. Kobow and colleagues performed DNA methylation and copy number profiling in a series of 26 PMG and 63 reference samples to identify novel molecular diagnostic markers that help classification of MCD. The authors showed that PMG formed a distinct methylation class. Moreover, they identified a new subset of patients with a brain mosaic 1q whole arm duplication that presented clinically with a unilateral isolated PMG, early-onset focal epilepsy and developmental delay. While none of the clinical signs defining the PMG 1q group were unique, the combination of all appeared to be exclusive. Taken together, the results support an integrated molecular-genetic and histological disease classification of cortical malformations including PMG, suggesting that genetic findings may play an important role in how PMGs are defined in the future. Although not the focus of the study, it is interesting that 17/26 PMG patients had a post-surgical outcome of Engel class 1 five years after surgery. This is higher than the reported average in non-FCD MCD as reported recently in a retrospective series of 9,147 surgical cases. The data indicate that molecular diagnoses underlying PMG may not only help clarify etiology, but may also be helpful in understanding outcomes.
This post was written by Katja Kobow and Ingo Helbig. Katja is Associate Professor for Molecular Neuropathology at the University Hospital Erlangen. Her research focus is on the role of DNA methylation and other epigenetic mechanisms in epilepsy. Katja has also been a long-standing member of the EuroEPINOMICS community and has previously contributed to blog posts on DNA methylation and neuropathology.
Katja Kobow is Associate Professor for Molecular Neuropathology at the University Hospital Erlangen, Germany. Her research focus is on the role of DNA methylation and other epigenetic mechanisms in epilepsy.
