
WGS. Whole-genome sequencing is increasingly used to understand the cause of rare diseases in a research and diagnostic context. However, while the usefulness of this technology has been shown in smaller studies, it remains unclear whether strategies to understand the cause of rare disorders through whole genome sequencing can be performed on a national level. A recent study in Nature reported the first results from a national sequencing campaign for rare disorders in the UK, including the analysis of more than 13,000 genomes. In this blog post, I would like to focus on the neurogenetics component of this enormous study, which identified disease-causing variants in GNAO1 as the most common cause within the study’s subgroup of neurological and developmental disorders.
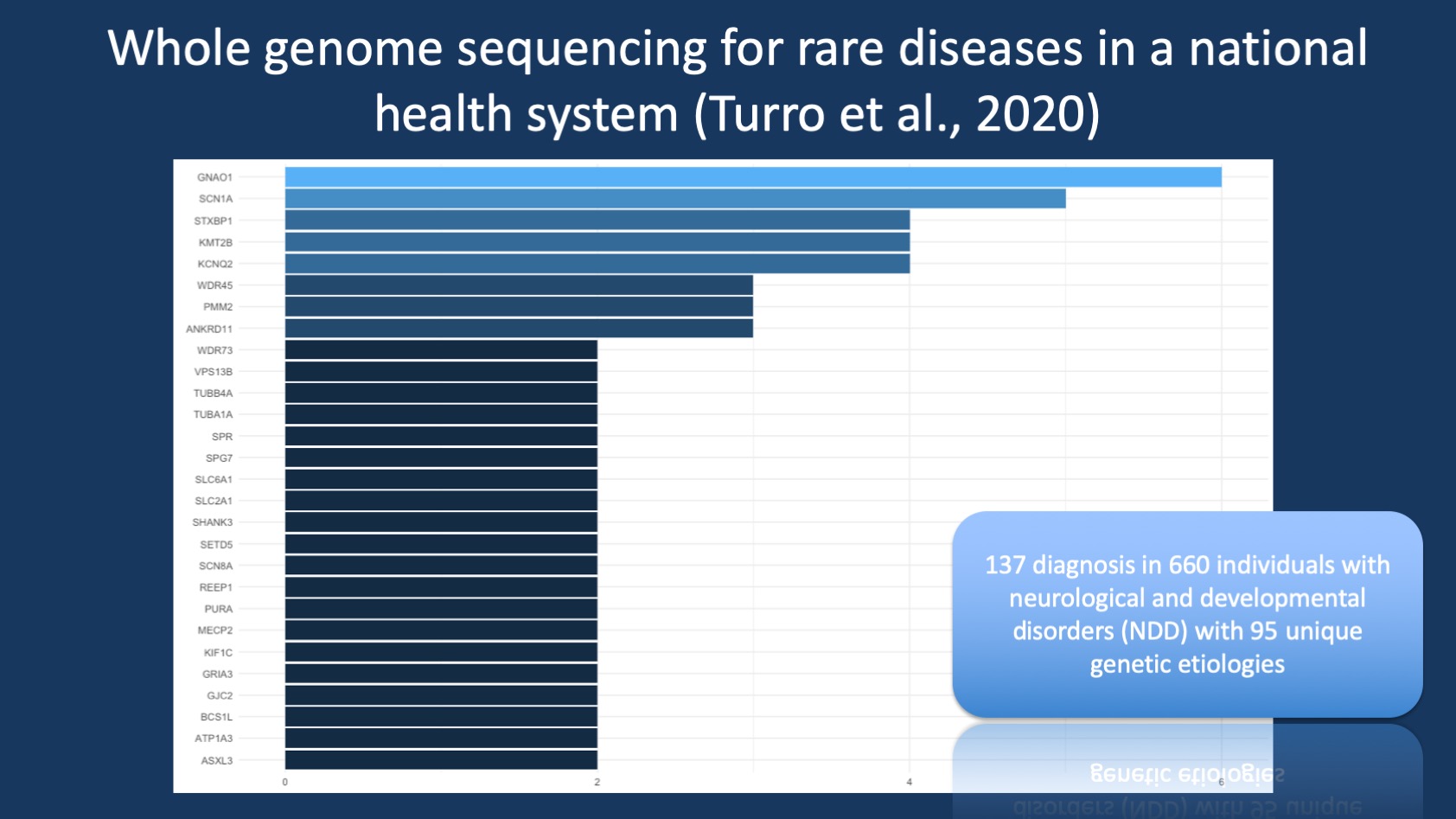
Figure 1. Frequency of genetic etiologies in the neurological and developmental disorders (NDD) subgroup of the study by Turro and collaborators, grouped by frequency and listing the genetic etiologies seen twice or more. Disease-causing variants in GNAO1, a rare neurogenetic condition causing a developmental and epileptic encephalopathy (DEE) and/or movement disorder, were the most common single-gene cause for neurological and developmental disorders, followed by SCN1A, STXBP1, and KMT2B.
Journal Club. This week, we held the first journal club for the research groups of our new epilepsy genetics program (ENGIN). We chose the publication by Turro and collaborators for this premier for a reason. Given that whole-genome sequencing is on the horizon for both clinical and research applications, this first study to assess the role of whole-genome sequencing on a national level was a good opportunity to glimpse at the future of neurogenetics. Whole-genome sequencing is often considered an ‘ultimate technology’ with the inherent promise that it would allow for discoveries far beyond what we can currently achieve. However, looking back at the last few years in neurogenetic research, this technology has not yet fully delivered this promise, and the advantage of whole-genome sequencing over whole-exome sequencing with regards to the diagnostic yield is incremental at best. However, if applied across a large number of rare diseases on a national level, what would such a technology look like?
100,000 Genomes. The study of Turro and collaborators was performed in the UK, where many large-scale genomic studies have emerged over the last decade, funded both by national efforts and non-profit organizations. In its early stages, EuroEPINOMICS benefitted enormously from the sequencing efforts by the Wellcome Sanger Institute and more recently, the Deciphering Developmental Disorders (DDD) study has pioneered trio exome sequencing at scale. On a national level, the UK has established the 100,000 genomes project through Genomics England, a national project sequencing whole genomes from National Health Service (NHS) patients. In the study by Turro and collaborators, 13,037 individuals with rare disorders underwent whole-genome sequencing; individuals were enrolled at 57 NHS hospitals at the UK and 26 hospitals in other countries. The individuals with rare disorders were assigned to various disease domains, with NDD (neurological and developmental disorders) representing one of these domains with 660 individuals. In addition, almost 5,000 individuals with rare disorders were not assigned to a disease domain, but enrolled in a pilot rare disease sequencing project.
Genomic diagnosis. With respect to the genomic diagnosis, a multi-disciplinary panel developed a list of diagnosis-grade genes (DGG) for the various disease domains. The number of candidate genes for neurological and neurodevelopmental disorders included 1,423 genes, far outnumbering the number of DGGs in other domains. This reflects the heterogeneity of the neurodevelopmental disorders with respect to genetic causes. In addition, I had two comments regarding the gene selection. First, I was surprised that the multidisciplinary panels developed a separate framework for selecting relevant genes, including a separate ‘language’ to refer to them. When we look at genes for neurodevelopmental disorders, there is an established framework to assess gene validity as per ClinGen criteria, and it was not clear from the publication to what extent the DGGs have been vetted by formal criteria versus expert opinion. Secondly, the list of genes for DGG seem excessive and includes genetic etiologies that are no longer considered disease genes for neurodevelopmental disorders, including EFHC1 and SCN9A. While these genes did not result in diagnoses in this cohort, I was a bit surprised to see that these etiologies withstood vetting by an expert panel for gene selection. Using this list of candidate genes, a ‘virtual gene panel’ was performed on the 660 individuals with NDD.
GNAO1. The most common genetic etiology identified in individuals with NDD was GNAO1, which was found in six individuals in their study. GNAO1 is a recently identified rare cause of developmental and epileptic encephalopathies and may also cause a severe childhood-onset movement disorder. The genetic diagnosis of SCN1A was made in five individuals, STXBP1-related disorders, KMT2B-related disorders, and KCNQ2-related disorders were identified in four individuals, respectively. The identified diagnoses allow for some conclusions with regards to patients who were primarily recruited into this study, namely individuals with epilepsies (SCN1A, KCNQ2) and movement disorders (KMT2B, GNAO1). It should also be noted that the cohort of individuals with NDD in this study is relatively small and that this distribution of genetic etiologies may not necessarily reflect the neurodevelopmental disorders in general. For example, in the 2017 DDD study with more than 4,000 individuals undergoing exome sequencing, only STXBP1 and KCNQ2 were amongst the most 20 most common genetic etiologies. The most common genetic causes in the DDD study, such as disease-causing variants in ARID1B, ANKRD11, or DDX3X were not found in a least two individuals in the study by Turro and collaborators. This demonstrates that there may be some inherent bias in some of the studies of neurodevelopmental disorders that allow for different genetic etiologies to become more prominent.
Comparison with other rare disorders. Compared with other rare genetic disorders in the study by Turro and collaborators, the neurodevelopmental disorders stood out in two different ways. First, the diagnostic yield was much higher (~33%) than in most other disease groups. Only the inherited retinal disorders had a higher diagnostic yield (54%), while most other disease groups yielded between 5% and 20%. This is in line with what we would expect in neurogenetics, even though many studies, especially lab-based studies in non-selected patient cohorts, have lower diagnostic yields. Secondly, the number of diagnostic reports per gene in the NDD was much smaller compared to some other disease groups. For example, for pulmonary arterial hypertension (PAH), more than 150 reports were issued for individuals with disease-causing variants in BMPR2, and more than 50 reports were issued for individuals with inherited retinal disorders caused by disease-causing variants in ABCA4. BMPR2 variants are known to be the cause of ~20% of PAH, but PAH is a relatively rare disease. Therefore, it is very informative to see how various rare disorders compare with regards to their genetic heterogeneity.
What you need to know. Over the next 2-5 years, whole genome sequencing will likely become a standard technology in a clinical and diagnostic setting. The study by Turro and collaborators examines the impact of this tool when applied to rare diseases across an entire health system. With respect to neurodevelopmental disorders including the epilepsies, the authors find a comparable diagnostic yield to prior studies, suggesting that a uniform genome sequencing combined with a ‘virtual gene panel’ approach is feasible. Analysis of non-coding regions, once thought to be the primary strength of whole genome sequencing, will likely remain a challenge for the near future. In this blog post, I was only able to highlight some aspects of the study by Turro and collaborators, which represents one of the most comprehensive studies in rare disease genetics to date.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
