
Pathogenic or benign. In 2018, ICK, coding for Intestinal-Cell Kinase, was reported as a novel causative gene in Juvenile Myoclonic Epilepsy (JME) in the New England Journal of Medicine. JME is one of the most common epilepsy syndromes, and the authors suggested that up to 7% of JME in their study may be explained by pathogenic variants in this gene, suggesting that, if applicable to all individuals with JME, it may provide a genetic diagnosis for an expected 500,000 individuals worldwide. In a reply to the initial study, the investigators of the EuroEPINOMICS-CoGIE, EpiPGX, Epi4K, and EPGP Consortia attempted to replicate these initial findings, but could not find any evidence in for a role of ICK in JME and indicated that the initial results may have arisen by chance and due to methodological issues. Given the potential implications for future research and therapy development in a relatively common epilepsy, the controversial ICK story is a good example to highlight why it is important to revisit the current consensus on when we consider a candidate a true disease gene and why a category mistake confusing variant pathogenicity for gene validity may result in false positive findings.
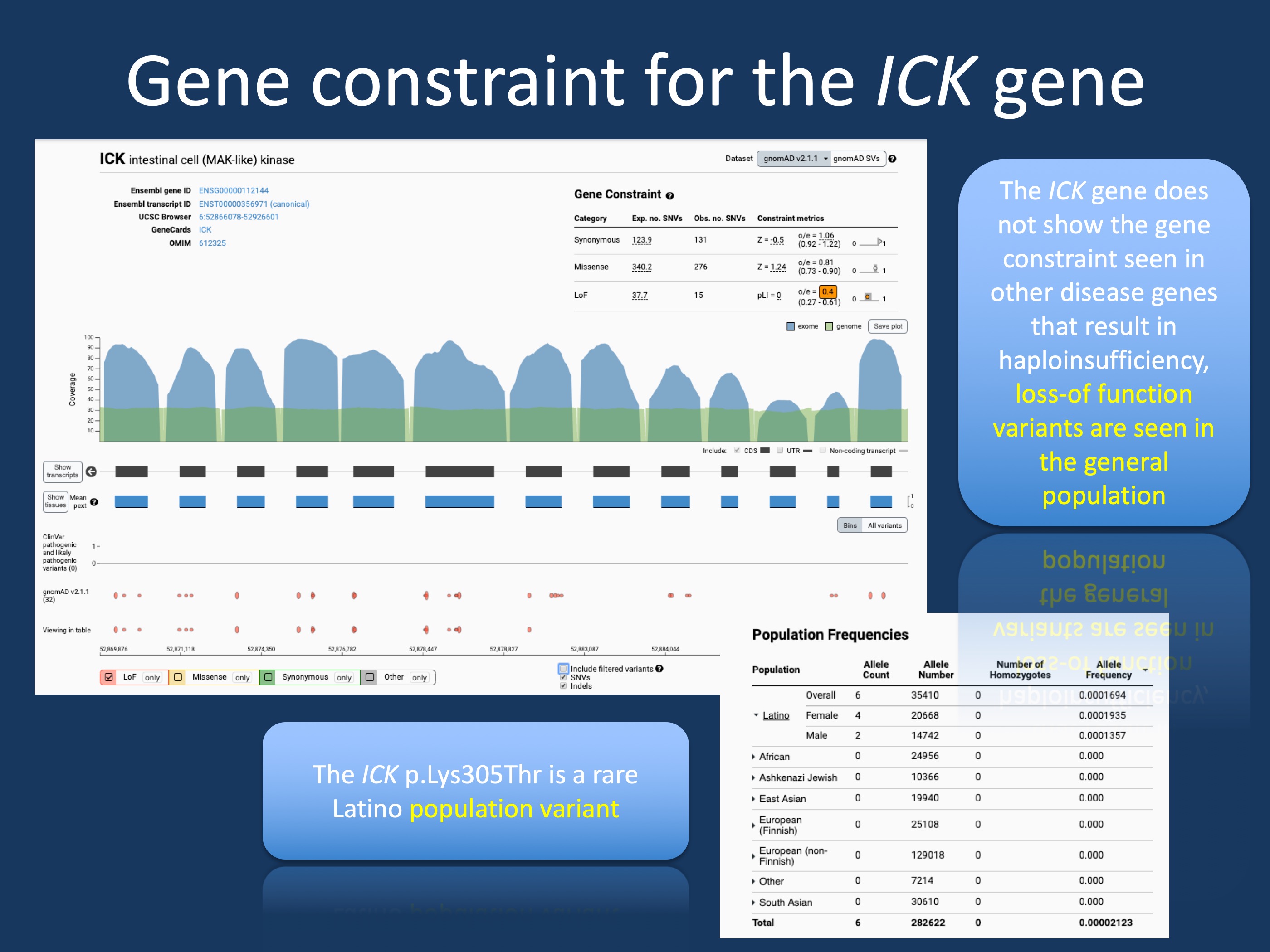
Figure 1. The population genetics of ICK and the c.914A>C (p.K305T) variant. The ICK gene is not constrained in the general population, i.e. loss-of-function variants are tolerated in the general population, which makes haploinsufficiency as a disease mechanism unlikely. The ICK c.914A>C (p.K305T) variant is a rare Latino population variant seen in 1:5000 individuals. This makes it unlikely that this variant is a monogenic disease variant causing familial Juvenile Myoclonic Epilepsy (JME).
Epilepsy genes. Let me preface this blog post by the statement that I am not neutral, as I am both a member of the EuroEPINOMICS consortium, authoring the letter to the NEJM editor and a member of the ClinGen Epilepsy Gene Curation Expert Panel, which is tasked with evaluating gene-disease relationships in the epilepsies. As of April 2019, we have not yet formally reviewed the evidence for or against ICK, so this blog post is not written in my role as the ClinGen Epilepsy Expert Panel leader. In addition, rather than taking sides in the discussion regarding ICK, I would like to lay out the general framework for when we call a gene a valid “epilepsy gene”, assessing a concept referred to as gene validity.
Gene validity versus variant pathogenicity. There are two major concepts that we discussed in our 2018 Primer of the Genetic Literacy Series: clinical validity of the disease-gene relationship and variant pathogenicity. Clinical validity of the disease-gene relationship asks whether there is sufficient evidence that support the role of pathogenic variants in a gene in a given disease. Variant pathogenicity assesses the evidence for a particular variant to be disease-causing. For example, a truncating variant in SCN1A in an individual with Dravet Syndrome is pathogenic as the gene is a valid disease gene for Dravet Syndrome (i.e. the clinical validity of the disease-gene relationship of SCN1A and Dravet syndrome is established) and haploinsufficiency is an established disease mechanism with absence of protein-truncating variants in the general population (variant pathogenicity: check). However, variant pathogenicity is irrelevant if the disease-gene relationship is not clinically valid. For example, you may find a de novo truncating variant in the Mucin 12 gene (MUC12) in an individual with Dravet Syndrome that has similar variant properties. However, as MUC12 is not a disease gene, this variant is not disease causing. In fact, MUC12 is a rather variable gene in the human genome with more protein-truncating variants than you would expect. Taken together, the validity of the disease-gene relationship must be established before assessing whether a given variant can be disease causing.
The ICK gene. In their 2018 publication, Bailey and collaborators initially identified an ICKc.914A>C (p.K305T) missense variant in a large family with JME including 12 affected individuals. The variant segregated within this family. They subsequently identified further missense variants in 22 of 310 additional patients from the GENESS consortium that has study sites in United States, Mexico, Honduras, Brazil, and Japan. While JME is a relatively mild phenotype, disease-causing variants are typically not found in unaffected individuals. However, the ICK c.914A>C (p.K305T) missense variant is found in allegedly unaffected Latino individuals in the gnomAD browser (Figure 1). While it is theoretically possible that such a scenario occurs, such a variant would usually not be considered disease-causing unless there is further evidence for this particular variant. A similar situation is encountered with the PRRT2 gene, where the common disease-causing c.649dupC variant is encountered in single individuals in large population databases (link), but the genetic evidence for this particular variant is overwhelming and confirmed over time. This is conceptually different from the ICK c.914A>C (p.K305T) variant. Accordingly, even a low population frequency is typically a strike against a variant being disease-causing and suggests that the ICKc.914A>C (p.K305T) variant is a population variant. If this variant that is seen in 1:5000 individuals were a true disease variant, it alone would account for 20% of all JME. Basically, genetic variation has been recognized as so prevalent that genes have fight an “uphill battle” to convince us that they are disease causing. This evidence needs to occur primarily on a genetic level, e.g. functional data cannot compensate for the lack of genetic evidence.
ICK in the general population. If we hypothesize that ICK variants are disease-causing through a loss-of-function mechanism, we would typically expect protein-truncating variants to be very rare or absent in the general population, a measure that is quantified by the probability of being loss-of-function intolerant (pLI) score that is implemented in the gnomAD browser. Genes such as SCN1A or STXBP1 have a pLI of 1.00, suggesting that loss-of-function variation is not tolerated in the general population. In other words, there is significant gene constraint. For ICK, the situation is different (Figure 1). The ICK gene is not constrained in the general population.
Validation. While is it possible that ICK only causes disease due to a low frequency population variant in a single family, such an isolated phenomenon usually would not account for sufficient gene validity and independent validation is required. This is what has been attempted in the joint EuroEPINOMICS-CoGIE, EpiPGX, Epi4K, and EPGP letter to the NEJM editor, which was not able to validate the initial results. While negative results do not “harm” the status of ICK as a rare disease gene in a single family, it demonstrates that this finding is not generally applicable. Where does this leave us with regards to evaluating the role of ICK in JME? Population genetics argues against haploinsufficiency as the disease mechanism and suggested pathogenic variant in the initial family is a rare Latino population variant. While there is significant genetic heterogeneity in the epilepsies that would allow for such a scenario to occur for true disease-causing variant, it is highly unusual in 2019 for a true gene-disease association to fail replication given the range of resources available. Past experience has shown that the alleged disease variants turned out to be benign population variants.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
