
HCN1 update. Hyperpolarization-activated cation channels (HCN) are involved in neuronal pacemaker activity and regulate neuronal excitability through hyperpolarization-activated Ih current. In 2014 de novo missense variants in HCN1 were identified in five unrelated individuals with a Dravet Syndrome-like developmental and epileptic encephalopathy (DEE). However, in the intervening four years relatively little additional evidence has emerged regarding the role of HCN1 in epilepsy. Now, a recent publication in Brain identifies additional individuals with HCN1-related epilepsies and significantly expands the clinical spectrum beyond Dravet-like DEE.
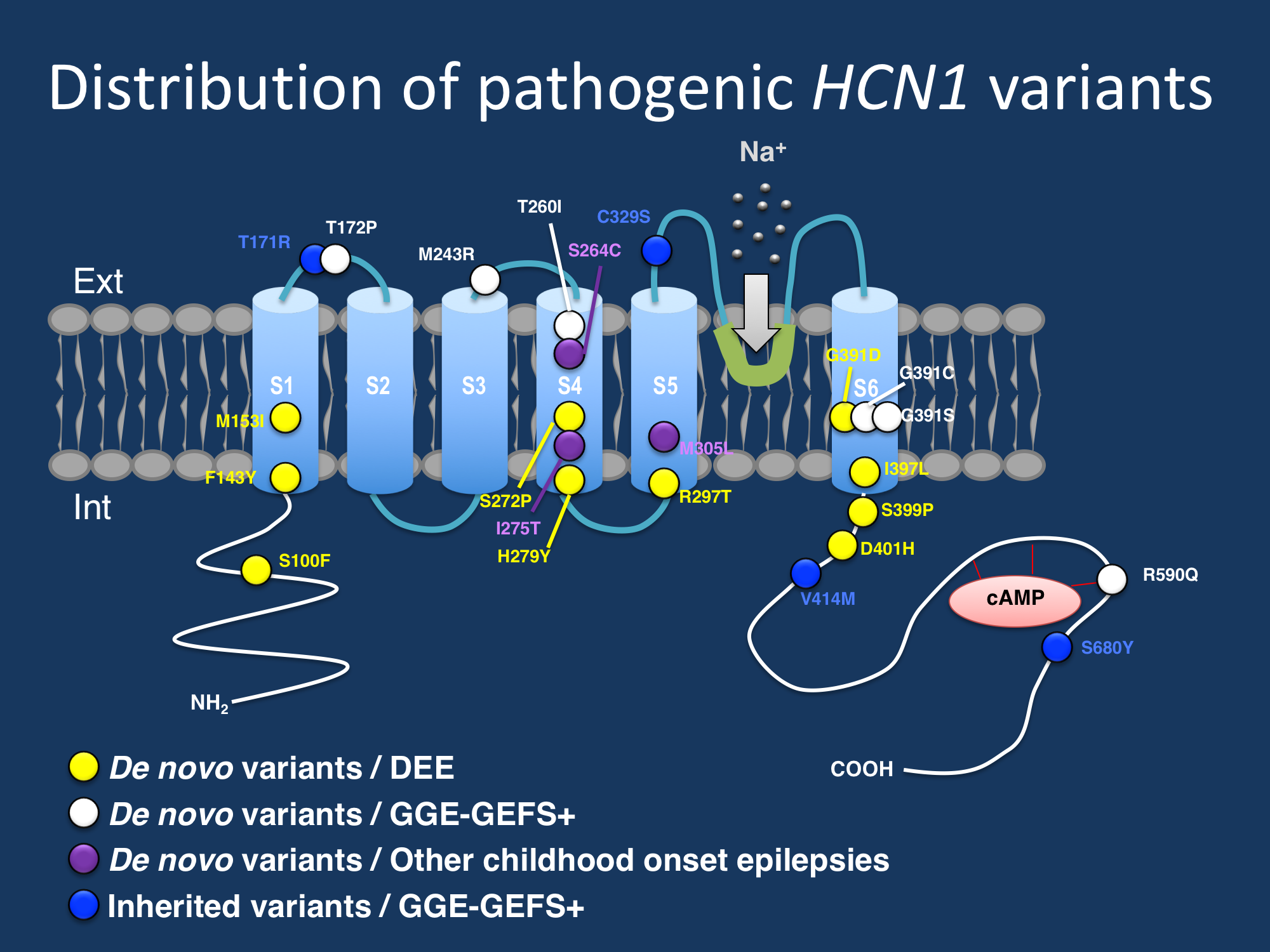
Figure. Distribution of identified variants across the HCN1 channel. De novo variants, particularly in patients with more severe phenotypes, cluster in S4, S5, and S6 segments, which form the voltage sensor and pore forming region of the channel.
The spectrum of de novo variants. In their recent publication,Marini and collaborators describe 19 newly identified individuals with de novo missense variants in HCN1. The clinical spectrum associated with de novo HCN1 variants is much broader than the developmental and epileptic encephalopathy originally described and surprisingly includes milder epilepsies within the GEFS+ spectrum. A wide range of seizure types were observed, including generalized tonic-clonic, clonic, atonic, tonic, absence, myoclonic, and focal seizures. Fever-associated seizures were common, seen in over 60% of patients. Developmental outcomes were unexpectedly broad, ranging from severe intellectual disability to normal cognitive outcome. Observed epilepsy syndromes within the de novo group included febrile seizures plus (FS+), IGE/GGE and other generalized epilepsies, and epileptic encephalopathies including epilepsy of infancy with migrating focal seizures. Although the phenotypes among sporadic patients was extremely heterogeneous, there was some clinical concordance among individuals with the same variant. For example, two patients with the p.Gly391Asp variant both had neonatal-onset epileptic encephalopathies, daily refractory seizures, and severe cognitive impairment. Two patients with a different amino acid substitution affecting the same codon p.Gly391Ser had FS+ and normal cognition to mild ID with infrequent seizures. These findings suggest that the phenotype may be associated with the underlying genetic variant
Familial HCN1 variants. Four small GEFS+ families were identified with missense HCN1 variants that segregated with the phenotype. All affected individuals in these families had epilepsy phenotypes within the GEFS+ spectrum, including FS+ and other generalized epilepsies with normal cognitive outcomes. However, six individuals carrying the familial HCN1 variant did not have epilepsy, suggesting that HCN1 variants in families are not fully penetrant and that more research is required to understand the consequences of inherited HCN1 variants.
Functional analysis. Whole-cell patch clap analysis of several de novo and familial HCN1 variants in CHO cells showed highly heterogeneous results, including loss-of-function and gain-of-function properties such as reduced current density, right shift of voltage-dependent activation, and changes in channel gating kinetics. These results indicate that it is not possible to categorize a functional mechanism for HCN1-related neurodevelopmental disorders as broadly “loss-of-function” or “gain-of-function” and that the functional effects are likely highly variant specific.
What you need to know. In their recent publication in Brain, Marini and collaborators publish an update on HCN1-related epilepsies, which significantly expands the phenotypic spectrum beyond Dravet-like developmental and epileptic encephalopathies. Nearly 40% of individuals with de novo HCN1 variants develop severe epilepsy and cognitive impairment consistent with developmental and epileptic encephalopathy. However, another 40% of identified patients – both sporadic and familial – present with milder fever-sensitive generalized epilepsies and with mild intellectual disability or normal cognition. These findings suggest that pathogenic variants in HCN1 also can cause phenotypes within the GEFS+ spectrum, with a very wide clinical spectrum that may be related to the underlying genetic variant. Functional consequences of pathogenic HCN1 variants are highly variable and variant specific, and can include either gain- or loss-of-function properties.
Thanks for the update. It seems to be a general observation that ion channel mutations can result in a large clinical spectrum, depending on the functional effect specific to each mutation, and even differing for different substitutions at the same residue. It is not surprising, considering the complexity of ion channel function and the evolutionary conservation of many residues from invertebrates to human.
Yes, I absolutely agree with you that, really, it should be surprising if we don’t see a clinical spectrum associated with variants in a particular ion channel-encoding gene! It speaks to the fascinating nature of ion channel physiology.