
The X-factor. Interpreting variants in X chromosome genes in a clinical context is an ongoing diagnostic challenge, regardless of whether the variant is identified in a male or female patient. The majority of X-linked conditions affect hemizygous male individuals, with heterozygous carrier girls and women largely unaffected or much less severely affected. PCDH19-Epilepsy is, of course, a notable rule breaker in this regard. However, we are learning that other X-linked conditions don’t play by the traditional rules either, and affected heterozygous females are being described for some other X-linked conditions. In some cases, including SMC1A– and NEXMIF– (formerly called KIAA2022) related disorders, the phenotypes in males versus females are more or less distinct. However, in other X-linked conditions, including IQSEC2-encephalopathy, both affected males and females share a continuum of similar features. A recent publication in Genetics in Medicine explores and expands the spectrum of IQSEC2-encephalopathy and delves into what is similar – and what is distinct – in affected male and female patients.
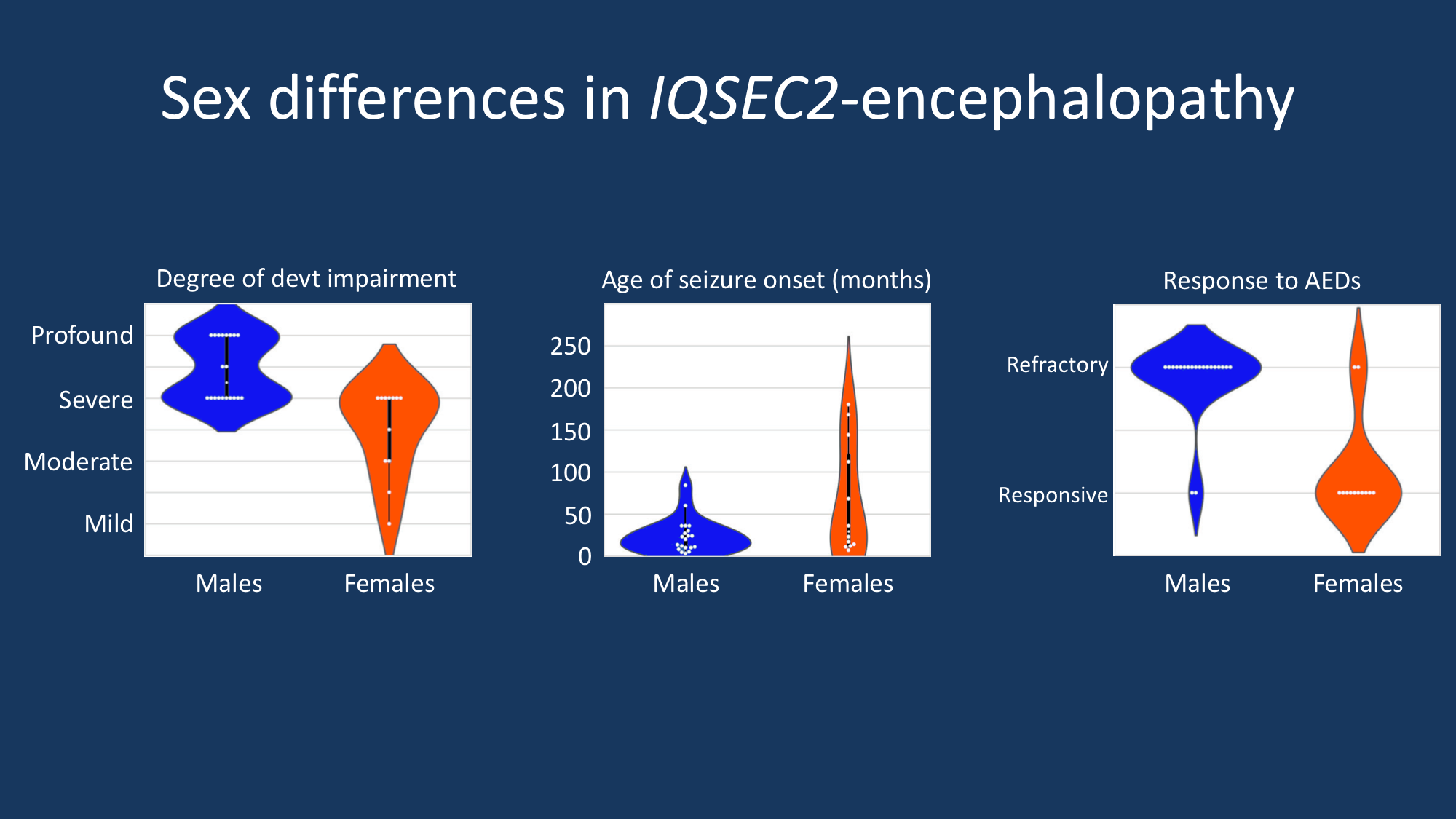
Although both males and females can be affected by this X-linked condition, specific phenotypic differences emerged between males and females with IQSEC2-related encephalopathy. Affected males were found to have more severe developmental impairment, earlier seizure onset, and poorer response to antiepileptic drugs compared to affected females. The phenotype in affected females is more variable, with a broader range of developmental outcomes, age of seizure onset, and response to anti-epileptic drugs (AEDs).
An X-linked disorder of both sexes. The current study by Mignot and collaborators explores the genetic landscape and phenotypes in 37 newly described patients with IQSEC2-encephalopathy, greatly adding to our understanding of this genetic neurodevelopmental disorder. The cohort includes 18 male patients and 19 female patients, indicating that this is a disorder that affects both sexes with equal frequency. The overwhelming majority of IQSEC2 variants (88%) in both male and female patients are loss-of-function and lead to premature termination codons. All affected females had de novo variants. Variants were de novoin 75% of affected males but were maternally transmitted in 25%. Although boys and girls are affected with similar frequency, the severity of disease generally appears to be more severe in males compared to females, with more severe developmental and seizure outcomes in boys compared to girls. Of the patients with milder developmental outcomes, all were female.
The IQSEC2-phenotype. Epilepsy is a common feature in both male and female patients with IQSEC2-encephalopathy, although it is more common in boys than girls. Of the affected female patients, 70% had epilepsy, whereas 95% of affected males had epilepsy. Median age of seizure onset was 34 months. Of patients with epilepsy, generalized seizure types are predominant, including atypical absence, generalized tonic-clonic, tonic, atonic, and myoclonic seizures. Epileptic spasms are also common. Development is severely impacted, with the majority of individuals having severe to profound impairment. Delay is typically more severe in males than females; for example, females are more likely than males to acquire independent walking and speech. Behavioral issues are also prominent, including autistic features, hyperactivity, and self-injurious behavior.
What does the IQSEC2 do? The IQSEC2 gene was identified in human neurodevelopmental disorders through genetic studies. The overwhelming genetic evidence suggests that the biological pathways that this protein is involved in must be critical for neurodevelopment. Interestingly, some of the biology of the IQSEC2 protein had already been elucidated before it became a target in genetic studies – at the time IQSEC2 was referred to as BRAG1. The IQSEC2/BRAG1 protein is a guanine nucleotide exchange factor found in the post-synapse of excitatory neurons and serves as a regulator for GTPases. IQSEC2/BRAG1 is important for trafficking in the postsynapse, including the regulation of glutamate receptors. Therefore, by playing an important role in the plasticity of the postsynapse, the IQSEC2/BRAG1 protein is located at a critical junction where synaptic signaling is integrated. This provides a framework to understand why pathogenic variants in IQSEC2 result in neurodevelopmental disorders.
What does X-inactivation have to do with it? In many X-linked conditions, the sex-specific differences in phenotypic expression come down to X-chromosome inactivation, also sometimes called lyonization after Dr. Mary Lyon, the English geneticist who first proposed the mechanism. Biological sex in humans is determined by our sex chromosomes, the X and Y. X-chromosome inactivation occurs early in female embryonic development and is a process by which one X chromosome is randomly silenced. All cells that derive from that cell inherit the same X-chromosome inactivation pattern, which is stable over time. Typically, girls affected with X-linked disorders have highly skewed patterns of X-inactivation, with the X chromosome carrying the pathogenic variant activated and the X chromosome carrying the unaltered copy of the gene silenced. Although most genes on the X-chromosome are subject to this epigenetic silencing, some genes escape inactivation and are expressed from both X chromosomes. IQSEC2 happens to be one of these genes that escapes inactivation. This is supported by the observation by Mignot and colleagues that X-chromosome inactivation patterns in affected girls were highly variable and inconsistent. Most affected females actually had random X-inactivation patterns, and no correlation emerged between X-inactivation patterns and severity of disease. Therefore X-inactivation is likely not explaining the sex differences between boys and girls with IQSEC2-encephalopathy.
What you need to know. Although most X-linked conditions have typically been considered restricted to affected males, many X-linked disorders, including IQSEC2-encephalopathy, are now emerging that show features in both sexes. Mignot and colleagues present the largest study to date of IQSEC2-encephalopathy with 37 newly described patients. Males with IQSEC2-encephalopathy appear to be more severely affected in general than affected females, although both sexes are affected with similar frequency. Epilepsy beginning in early childhood, usually with generalized seizure types and epileptic spasms, is seen in a majority of affected boys, although 30% of affected girls do not have epilepsy. De novo loss-of-function variants are typically identified in affected individuals, although missense variants have been reported, and maternally inherited variants are observed in about one-quarter of affected boys. X-chromosome inactivation does not appear to be playing a role in affected females, and is therefore not useful in evaluating a potential IQSEC2 variant in an affected girl.