
Unsolved cases. We are in an era of dramatic progress in understanding the genetic causes of neurologic disorders. In spite of this progress, many cases remain unsolved even after whole exome sequencing. One hypothesis for this missing heritability is that “non-coding” mutations outside the exome may explain at least some of these unsolved cases. A recent study looked at de novonon-coding variants in patients with neurodevelopmental disorders. The study sheds new light on this question and reminds us that, despite all the recent progress, there is much still to learn about vast portions of the genome.
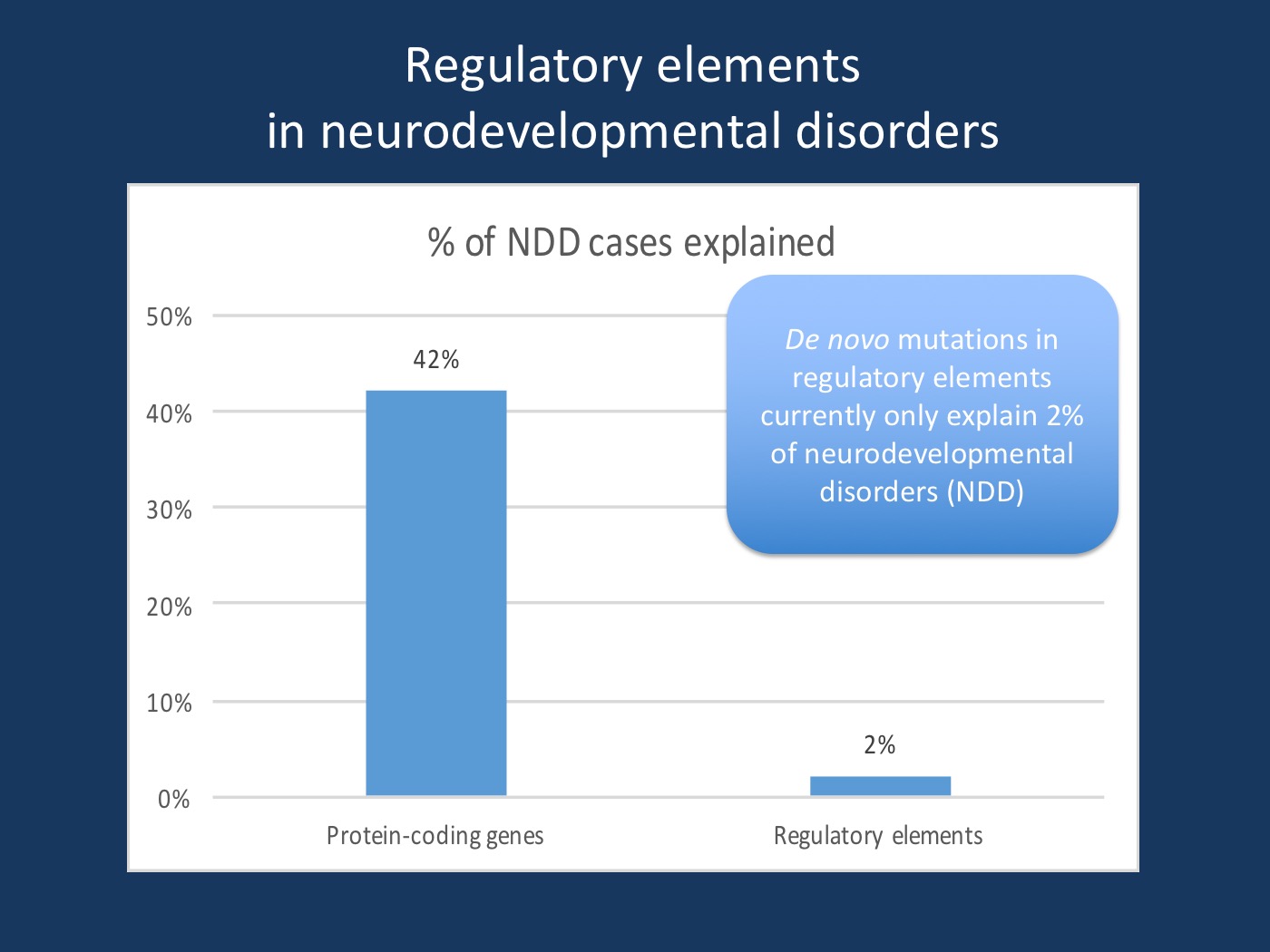
De novo mutations in coding versus non-coding variants. Although they do play a role in neurodevelopmental disorders, de novo variants in regulatory elements explain a much smaller number of cases (1-3%) than do protein-coding variants (42%), according to the recent study by Short and collaborators.
Regulatory elements. The Epilepsiome blog, like the field of genetics generally, has focused primarily on the exome – the genetic sequences that encode proteins. That is because 85% of the known monogenic causes of human disease involve protein-coding sequences. But protein-coding DNA makes up less than 2% of the entire genome. The remaining 98% of the genome is non-coding. This includes introns, repetitive DNA (such as LINEs, SINEs and satellite repeats), and other miscellaneous DNA sequences. Included among this non-coding DNA are regulatory elements, which help control gene expression and determine timing and quantity of protein produced. For example, promoters are where transcription factors bind to begin gene transcription into RNA, and lie immediately upstream of genes. Other regulatory elements, such as enhancers and silencers, can lie quite distant from the genes they regulate. They are thought to regulate expression when they are brought into proximity of a gene by bending of the DNA strands. It is reasonable to hypothesize that mutations in these regulatory elements would alter gene expression and contribute to human disease. However, the magnitude and the mechanisms of this effect remain largely unknown.
Non-coding variants in neurodevelopmental disorders. In a recent study, Short and colleaguesasked whether de novovariants in regulatory elements contribute to neurodevelopmental disorders. The study included more than 6,000 patients and their parents who participated the Deciphering Developmental Disorders (DDD) study. The authors sequenced DNA from introns, evolutionarily conserved regions, and two sets of putative enhancers. In total, the regions studied covered 4.6 Mb, which is approximately 0.1% of the non-coding genome. They compared the observed number of de novo variants in these regions to the number expected by chance based on baseline mutation rates. They found significantly more de novo variants than expected, though only in a specific subset of the regions they tested: evolutionarily-conserved regions with brain-specific activity. This suggests that conserved non-coding regions do play a role in neurodevelopmental disorders. However, the effect size was small: these variants were estimated to collectively explain 1-3% of the cases, compared to 42% explained by de novo variants in protein-coding genes.
The non-coding haystack. The study by Short and collaborators tried four differed methods to predict gene targets of regulatory elements, found poor concordance among the methods, and were unable to confidently link their regulatory elements with specific genes. This difficulty also limited their ability to cluster the regulatory elements by function or cellular pathway, as these functional links were unknown. This study demonstrates several aspects of why studying the non-coding portions of the genome is so difficult:
- We don’t always know where to look. Regulatory regions often are not located next to genes, and they don’t have characteristic sequence features to help identify them. This study had more success using evolutionary conservation as a guide than using lists of putative enhancers.
- Once we identify a regulatory element, which gene does it regulate? It is often not the closest gene, and there is no way to tell from the DNA sequence alone which regulatory elements link to which genes.
- It is difficult to predict the functional consequence of a non-coding variant. There is no equivalent of the triplet code, and no amino acid substitutions or stop codons, the features we use to predict the functional consequences of exonic variants. The study by Short and collaborators found that CADD scores did not correlate with evolutionary constraint and were not informative to identify deleterious non-coding variants.
- Non-coding regions of the genome are vast. This study was limited to de novo variants in highly conserved regions, covering 0.1% of the genome. Studying other non-coding variants without these constraints is likely to be even more challenging. If finding a disease-causing exonic variant is like finding a needle within a haystack of benign variants, then identifying a pathogenic non-coding variant is like finding a piece of hay that looks just like all the others, without knowing which features distinguish the piece you’re looking for.
What you need to know. At the moment, we are limited in our ability to study the non-coding genome and its role in human disease. The large, rigorous study by Short and collaborators found that regulatory non-coding elements do play a role in neurodevelopmental disorders, but the magnitude of that role was disappointingly small. Either this is not a major cause of disease or, more likely, we just don’t know how to find it yet.