
E2. When we work up a new-onset epileptic encephalopathy in clinical practice, there is a discrepancy between what we know and what we do. While we know that we have an almost 30% chance to find a causative de novo mutation in any of the known epilepsy genes, we usually think about a possible underlying inherited metabolic disorder when we order our first round of tests. However, the full phenotypic spectrum is often unknown and the question remained how many of these inherited metabolic disorders are missed. In our recent publication of the E2 consortium, we looked for evidence of inherited genetic disorders in patients with epileptic encephalopathies. Follow us on our journey that led to a negative answer, but uncovered a complexity in finding inherited diseases that we did not anticipate.
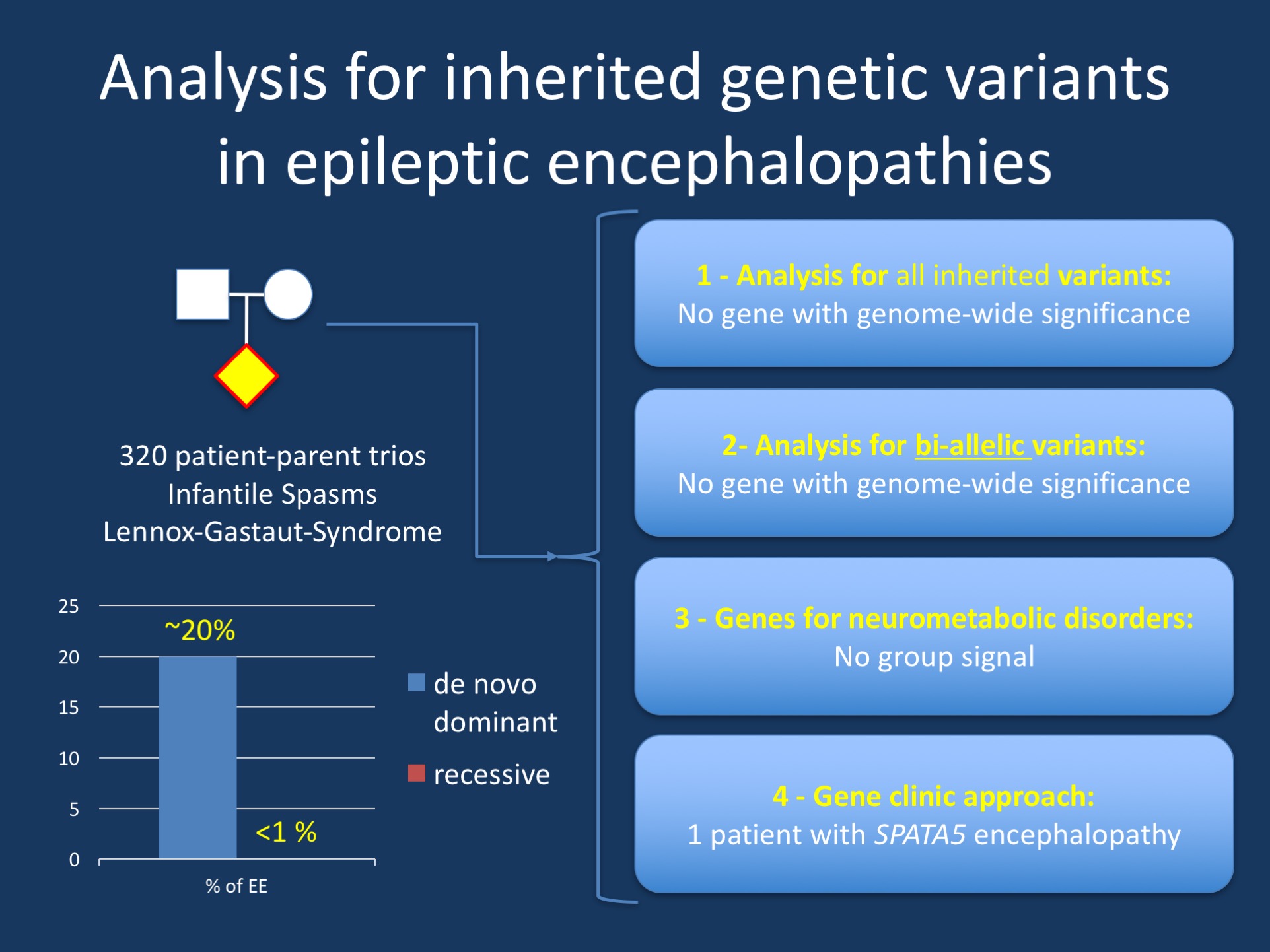
Figure 1. The most recent E2 publication assessed the role of inherited variants in patients with epileptic encephalopathies. While many genes for recessive disorders are known and while these genes can occasionally be identified in patients with epileptic encephalopathies, these genes are absent in the E2 cohort, a cohort of patients with epileptic encephalopathies that were generally pre-screened for metabolic disorders. There were no “hidden metabolic disorders” that were uncovered through genetic testing.
Transmission. In our recent E2 publication, we were interested in the role of inherited genetic variation in epileptic encephalopathies. Inherited genetic variation comes in different forms, ranging from a genome-wide signal in a specific gene to recessive monogenic disorders. Therefore, we followed the approach of testing four types of inherited variation in our cohort of 320 patients, starting with a genome-wide approach for any inherited variants, a genome-wide approach for genes with homozygous or compound heterozygous rare variants, and a specific test for 99 genes that are known to cause recessive disorders, including genes for mitochondrial disorders, neurotransmitter disorders, disorders of amino acid metabolism, urea cycle disorders, or disorders of fatty acid oxidation. Finally, we applied a “gene clinic” approach to examine inherited variation in each trio.
Negative findings. To make a long story short, all tests were convincingly negative and did not show any gene with genome-wide significance, no signal on a group level for genes in known metabolic diseases and – with a single exception – no relevant findings on the individual patient level. The only exception was a single patient with compound heterozygous variants in SPATA5, a recently identified gene for recessive epileptic encephalopathies, but also additional features such as microcephaly, dystonia, and hearing loss. The phenotype of the identified patient was fully consistent with the reported SPATA5 phenotype, strengthening the suspicion that this gene was causative.
Limitations. In brief, except for a single patient with SPATA5 encephalopathy, there is no evidence for inherited genetic variation in the E2 cohort. For clinical practice, this statement has two implications. First, there is no evidence that we are missing a large proportion of metabolic disorders in patients with epileptic encephalopathy. Patients in the E2 cohort were generally pre-screened for metabolic disorders, but given the international character of this cohort, this was not done in a systematic manner and mainly performed by the guidelines and practices of the local providers. This makes the E2 cohort a good cross-section of the overall patient population. Again, there is no evidence for hidden metabolic disorders in this cohort from a genetic perspective. Secondly, we realized that our current cohort is much too small to allow for any meaningful analysis for novel recessive genes. For example, in the roughly 3,500 genes that had at least one family where homozygous or compound heterozygous variation was observed, no single gene was significant. There are few interesting candidates in this list including SYNJ1, which is a known cause of autosomal recessive Parkinson Syndrome and epileptic encephalopathy, CEP120, a known gene for Joubert Syndrome, and KATNB1, which is the cause of a rare microcephaly syndrome. However, if there is a signal, it is hidden in the noise where we cannot say whether these findings are meaningful or simply by chance.
What does it mean? While we do not find evidence for known recessive disorders in patients with epileptic encephalopathies, we cannot conclude that the absence of evidence is in fact evidence of absence. In contrast to de novo mutations, where the E2 cohort was large enough to implicate several disease genes on a genome-wide level, there is too much inherited variation to tell signal from noise at this point. Larger sample sizes that will aggregate over time will allow for such an analysis to be performed in the future with sufficient statistical power.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
