
The sugar code. Many proteins in the human body undergo post-translational modification. A common mechanism to modify the function of proteins is a process called glycosylation, the adding of carbohydrate residues to protein. Glycosylation is probably the most complex post-translational modification, critically important to various physiological functions and therefore tightly regulated in cells. Accordingly, genetic disorders that interfere with glycosylation may present as severe, multisystem disorders. However, it is increasingly recognized that many congenital disorders of glycosylation have an exclusively neurological phenotype. Here is an update on ALG13 epileptic encephalopathy, a recently identified disease entity that may account for up to 2% of Infantile Spasms in females.
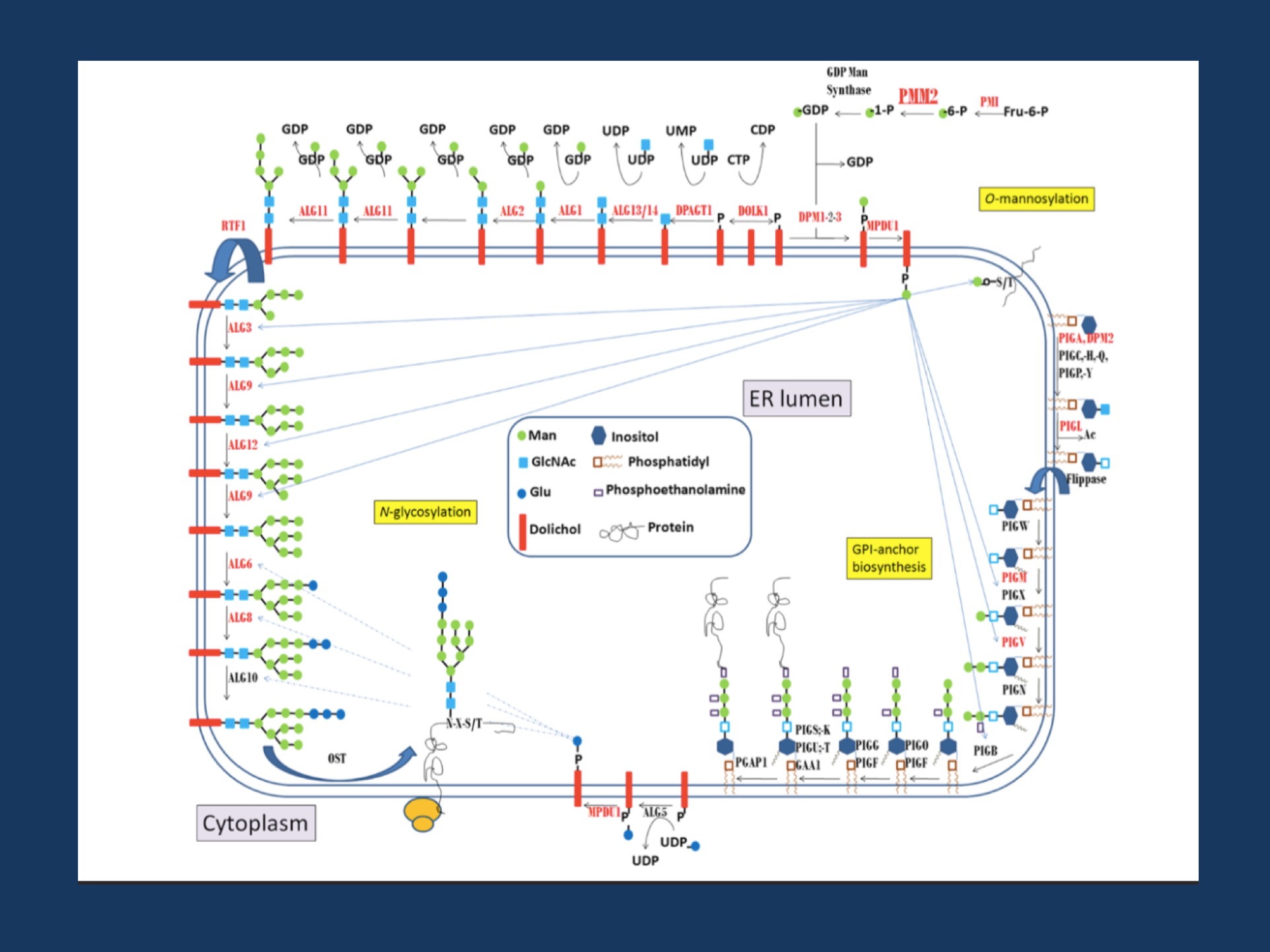
Biosynthetic pathways depending on mannose-1-phosphate: N -glycosylation, GPI-anchor biosynthesis, and O -mannosylation. Mannose-1-phosphate is the product of the phosphomannomutase activity, encoded by thePMM2 gene. The steps involved in the synthesis of the lipid-linked oligosaccharides required for the N-glycosylation of proteins, and glycosylphosphatidyl inositol required for the anchor of other proteins are detailed. The genes involved in each step are indicated, and those with mutations associated with congenital disorders of glycosylation are marked in red. Figure adapted from Morena-Bario et al 2013 GPI-anchor and GPI-anchored protein expression in PMM2-CDG patients. Orphanet Journal of Rare Diseases 8:170.
Epilepsiome page. This blog post should announce our ALG13 Epilepsiome page that was created by Elizabeth Dechene (CHOP Division of Genomic Diagnostics). We are currently reviewing the current genes related to human epilepsies in alphabetical order, building static reference pages for known epilepsy genes within the Epilepsiome project.
Biochemistry. Glycosylation is probably the topic where most of us nodded off in biochemistry class. The cell provides a plethora of mechanisms to add, modify and detach lengthy chains of carbohydrates to proteins. The most commonly used example of glycosylation is the ABO blood group system. It is the presence or absence of particular enzymes involved in glycosylation (glycosyltransferases) which dictates which blood group antigens are presented on red blood cells. Within the complex cascade of enzymes involved in correctly building the cellular sugar code, the ALG13 protein is involved in adding sugar chains to proteins in the endoplasmatic reticulum. Furthermore, glycosylation is important in the process of having a group of transmembrane proteins attached to the cellular membrane through GPI anchors. Impairment of this mechanism may lead to the clinical presentation of GPI anchor defects due to mutations in genes like PIGA, or PGAP2. We recently came in contact with this field by identifying a homozygous PGAP3 mutation in one of our families from the Israel-Palestine Epilepsy Family Project.
ALG13 related-CDG. The classical presentation of ALG13 deficiency illustrates some of the typical clinical features that are associated with Congenital Disorders of Glycosylation (CDG). Pathogenic variants in ALG13 are known to cause CDG Type Is, which is characterized by multiple systemic features including hepatomegaly, recurrent infections, and bleeding tendency. ALG13 is an X-linked gene and in affected boys, an atypical glycosylation profile can be observed, indicating a systemic dysfunction of glycosylation. This multi-system disease is hardly confused with the genetic epilepsies that we see in clinic. However, then this gene was virtually re-discovered.
ALG13 epileptic encephalopathy. When first exome studies were performed in patients with unexplained epileptic encephalopathy, a recurrent variant in ALG13 was identified in girls with intractable Infantile Spasms. Most patients identified so far carry the recurrent de novo p.N107S variant and in contrast to boys with CDG Is, the biochemical profile in affected girls in unremarkable. In parallel to other X-linked disease genes such as KIAA2022, there are discrete phenotypes in males and females. The mechanism why females have a phenotype predominantly characterized by epilepsy remains an active area of research. However, it is becoming increasingly clear that the phenotypic range of Congenital Disorders of Glycosylation expands into the group of epileptic encephalopathies.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
