
The story of TBC1D24. As with many epilepsy genes, the TBC1D24 story increases in complexity over time. Initially described to be associated with autosomal recessive familial infantile myoclonic epilepsy by Falace and colleagues and with autosomal recessive focal epilepsy by Corbett and colleagues in 2010, pathogenic variants in TBC1D24 have since been identified as a major cause of DOORS syndrome and have also been identified in individuals with familial malignant migrating partial seizures of infancy, progressive myoclonus epilepsy, early-onset epileptic encephalopathy, and autosomal dominant and autosomal recessive non-syndromic hearing loss. However, little is known about a potential genotype-phenotype correlation of TBC1D24-related disorders, as well as the underlying mechanism. Keep reading to learn more about recent discoveries related to TBC1D24.
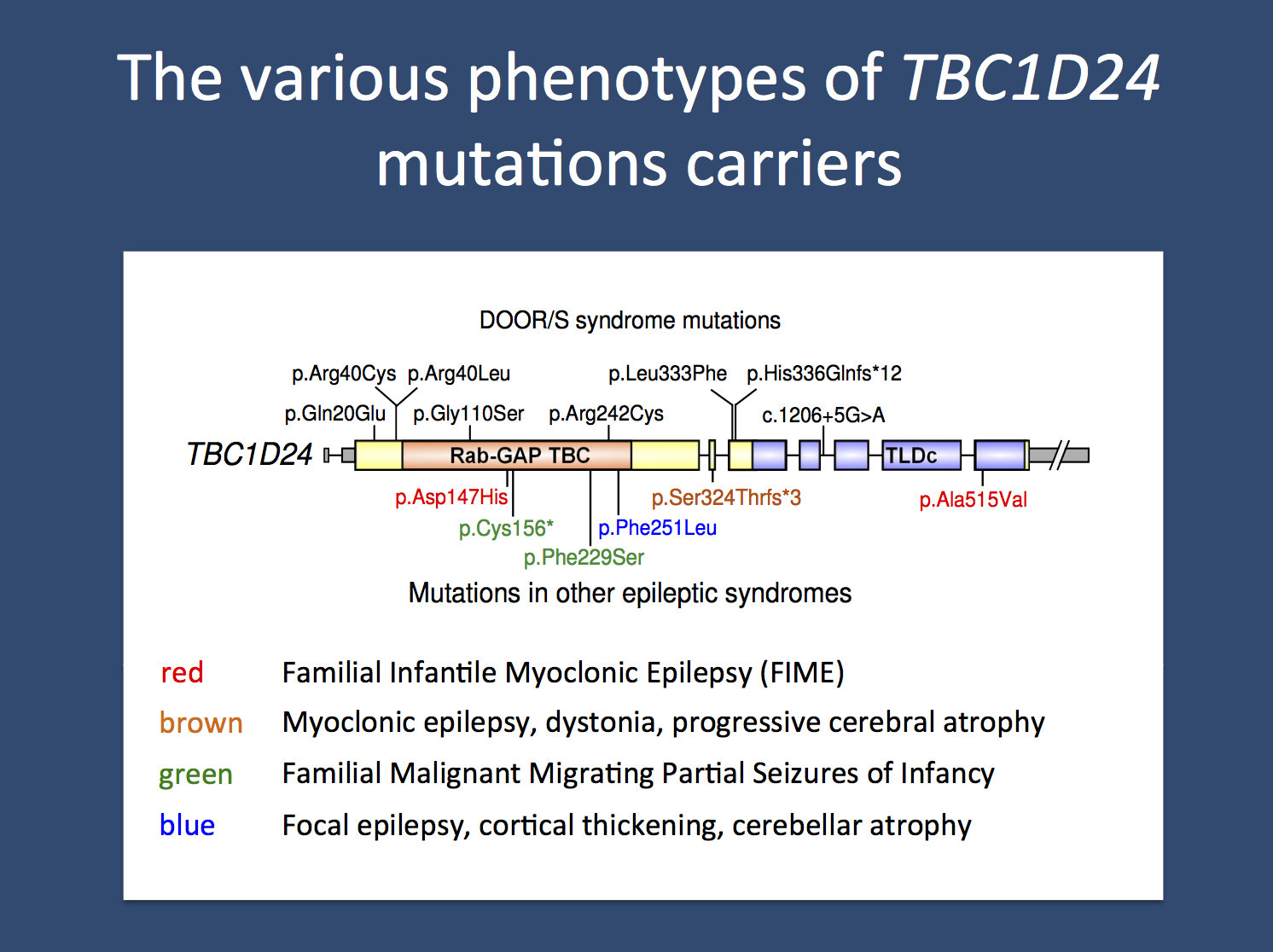
The various recessive and compound heterozygous epilepsy phenotypes due to mutations in TBC1D24.
What we are learning about TBC1D24. Our understanding of TBC1D24-related epilepsy continues to expand. Here are some of the highlights of what we have learned in recent months.
The continuing expansion of the TBC1D24 phenotype. Doummar and colleagues recently described a boy with TBC1D24-related epilepsy who helps illustrate the complexity of the TBC1D24 phenotype. The boy presented with multifocal attacks of myoclonus alternately affecting the hands, eyelids, limbs and sometimes the trunk, starting in infancy and occurring monthly. He did not present with other seizure types until about 8 years of age. Between 2 and 5 years, he developed episodic gait ataxia, mild cerebellar syndrome and intellectual disability. Brain MRI showed progressive hemispheric cerebellar atrophy with hypersignal of the cerebellar cortex and white matter.
TBC1D24 and protection against oxidative stress. Finelli and colleagues‘ recently published work suggests that disruption of the TLDc domain can lead to neurodegeneration. This domain is found in the OXR1 (Oxidation Resistance 1) gene, a gene that is important for neuronal cells’ resistance to oxidative stress. In addition, the TLDc domain is also found in none other than the family of proteins that includes TBC1D24. Finelli’s team found that expression of the TBC1D24 protein and other TLDc domain-containing proteins in dying neurons under oxidative stress led to a significant reduction in cell death, suggesting that these proteins can be neuroprotective. Mutant TBC1D24 protein containing a nonsense variant in the TLDc domain appeared to lose its neuroprotective qualities, supporting the hypothesis that this domain is important for protection against oxidative stress. Further studies are needed to understand how TBC1D24’s possible role in neuroprotection is related to its role in human disease
More TBC1D24 updates. Visit our TBC1D24 gene page to learn more about other updates on TBC1D24. Take a look at the additional topics that we have added with our revised gene page content, including sections on the clinical perspective, animal models, references, and research.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
