
GRIN1 encephalopathy. In the early 2000s Dalmau and collaborators observed a condition in women with ovarian teratoma who presented with psychosis or memory problems and rapidly progressing neurological deficits that required prolonged intensive care support. Auto-antibodies against the NR1 subunit of the NMDA receptor were found to be the causative agent. The clinical spectrum of anti-NMDA-receptor encephalitis has since expanded significantly and this initial discovery fueled the discovery of an entirely disease mechanism, the concept of the autoimmune encephalitis. In a recent publication in Neurology, we describe a novel neurodevelopmental syndrome affecting the gene for the NR1 receptor, the genetic sibling of NMDAR encephalitis. This blog post is about the unexpected overlap of autoimmune disorders with the genetic epilepsies and the spectrum of GRIN1-related genetic encephalopathies.
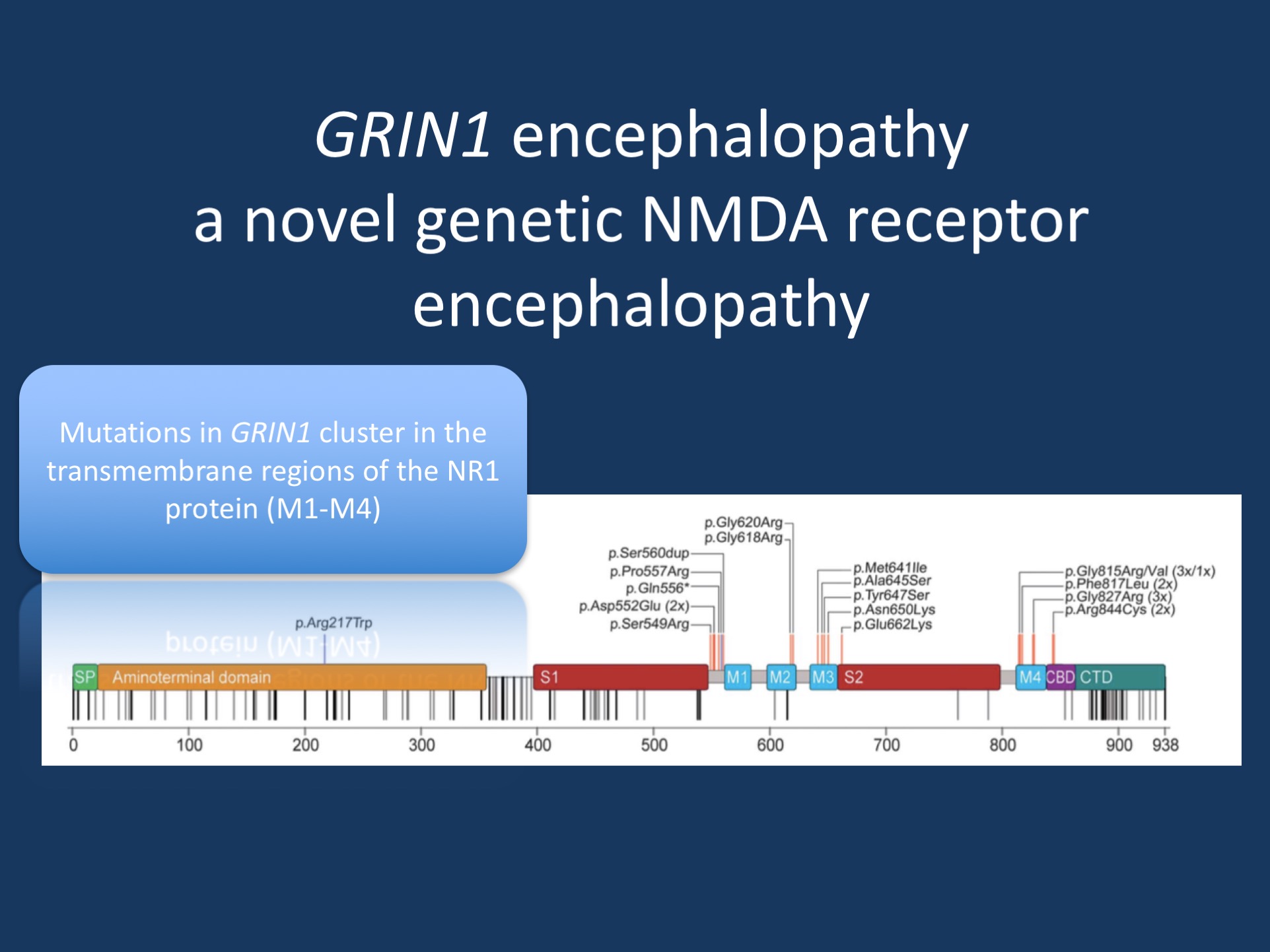
Figure 1. Signal peptide (SP), the extracellular N-terminal domain, and ligand binding sites (S1, S2), the transmembrane domains (M1-4), as well as the intracellular C-terminal domain (CTD) with the proximal Ca21 calmodulin binding domain (CBD). De novo mutations (red) cluster within or in very close proximity to M1-4. In addition, this region is particularly spared from nonsynonymous genetic variation according to the ExAC browser (rare/single variants, gray; repeated/frequent variants, black). The 2 homozygous GRIN1 variants are marked in blue. Adapted under a Creative Commons 4.0 License (CC BY-NC- ND), from Neurology.
The NMDA receptor. NMDA receptors are the one of the major receptors for the neurotransmitter glutamate in the Central Nervous System. With glutamate being the main excitatory neurotransmitter in the brain, NMDA receptors are a key component of the neuronal transmission apparatus. A typical NMDA receptor consists of four subunits, including two NR1 subunits and two NR2 subunits. Previously, we described pathogenic variants in GRIN2A (NR2A) and GRIN2B (NR2B) in genetic epilepsies. In our recent publication in neurology, we now identify the phenotypic spectrum associated with pathogenic variants in GRIN1, the gene coding for the obligatory main NMDA receptor subunit GRIN1.
From encephalitis to genetic encephalopathy. LGI1, CASPR2, and now GRIN1. At least three genes and their protein products have the following features: (a) mutations in these genes lead to genetic epilepsies and (b) autoantibodies against these proteins lead to a specific form of autoimmune encephalitis. Mutations in LGI1, for example, lead to a familial lateral temporal lobe epilepsy. Auto-antibodies against the LGI1 protein, in contrast, lead to a particular seizure type called faciobrachial dystonic seizures. Both the autoimmune condition and the genetic epilepsy differ significantly in their clinical picture – autoimmune conditions are acute conditions, often with a dramatic presentation, whereas genetic epilepsies are chronic, largely neurodevelopmental conditions. However, there are common features. For example, in both LGI1-related conditions, dysfunction of the temporal lobe is the neuroanatomical substrate. In our recent publication, we tried to contrast the presentation of GRIN1 encephalopathy (genetic) with anti-NMDA receptor encephalitis (autoimmune).
The GRIN1 encephalopathy spectrum. In our recent publication, we describe the phenotypic features of 23 patients with GRIN1 encephalopathy. Most patients had de novo pathogenic variants in GRIN1, but we also identified two families with homozygous variants in GRIN1, blurring the boundary between neurodevelopmental conditions due to de novo autosomal dominant and recessive inheritance. The pathogenic GRIN1 variants were found to cluster in the transmembrane segments of the NR1 protein. This resulted in a loss of channel function with a dominant-negative effect, explaining why both de novo variants and biallelic variants can be pathogenic. Heterozygous truncating GRIN1 variants, in contrast, are not pathogenic and can be found in population databases. This may be one reason why it took so long for GRIN1 encephalopathy to be recognized.
The phenotype. GRIN1 encephalopathy is a neurodevelopmental condition, often with severe intellectual disability and muscular hypotonia. Most patients identified in our cohort were non-verbal and never acquired the ability to walk. However, as with many other neurodevelopmental conditions, we are aware that this initial report may only describe a certain aspect of the disease phenotype and that we may get a better understanding with additional patients being recognized. In our series, approximately 70% of patients had seizures with various degrees of severity and a broad age of onset ranging from the first day of life to the age of 11 years. Interestingly, a significant proportion of patients also had a hyperkinetic movement disorder (~60%) and almost a quarter of patients had distinct oculomotor abnormalities such as oculogyric crisis. Typically, we don’t associate oculogyric crisis with neurodevelopmental conditions, but usually implicate neurotransmitter disorders, a subclass of neurometabolic diseases.
What you need to know. GRIN1 encephalopathy is an emerging entity, a neurodevelopmental syndrome due to de novo or biallelic pathogenic variants in GRIN1 that impair the function of the NR1 subunit of the NMDA receptor. GRIN1 encephalopathy has a broad phenotypic range and appears to have a more prominent movement disorder than other genetic NMDA receptor encephalopathies. The prominence of a hyperkinetic movement disorder, seizures, and sleep cycle dysregulation may be shared features of both autoimmune NMDA receptor encephalitis and genetic GRIN1 encephalopathy.
Open Access. I would like to thank Ambry Genetics for covering the costs for the Open Access fees for this publication, which allows the community to access the full text of this publication under a so-called Creative Commons 4.0 License. Ambry Genetics is the employer of my wife Katie who is one of the authors on this publication.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
