
An unexpected twist in the SCN8A story. SCN8A mutations were first implicated in epilepsy in 2012, when a de novo missense variant was identified in a patient with early infantile epileptic encephalopathy (EIEE) via genome sequencing. Since then, a number of patients with de novo heterozygous SCN8A variants and epilepsy have been reported, firmly establishing the role of SCN8A in EIEE, and we have learned a lot about the associated phenotype, mutation spectrum and disease mechanism within the last four years. Recently, a heterozygous familial SCN8A missense variant was identified in several families with a significantly milder epilepsy phenotype than reported in previous patients. Read further to learn more about the expanded SCN8A-associated epilepsy phenotype.
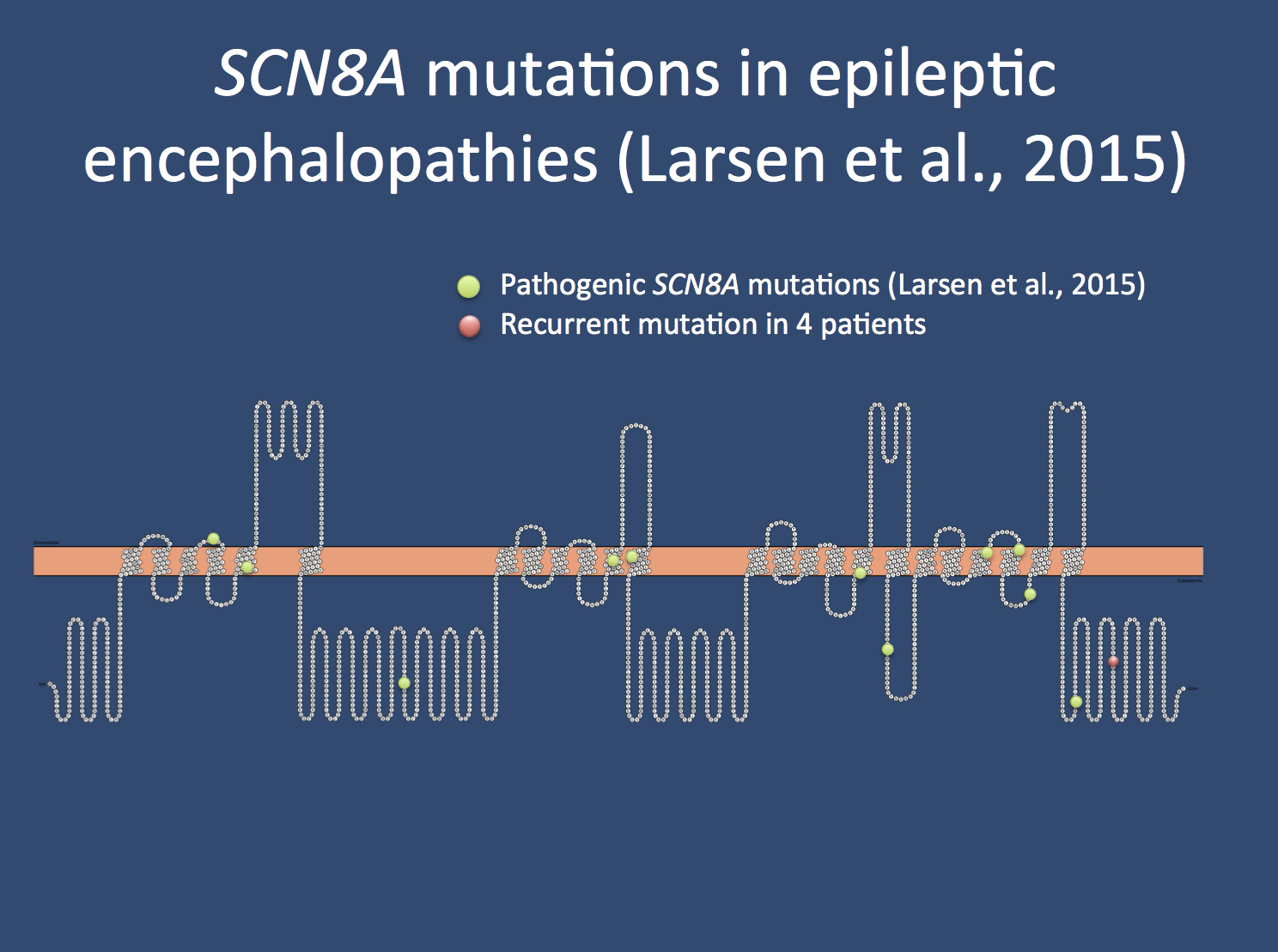
Spectrum of the SCN8A mutations identified by Larsen and collaborators.
Benign infantile seizures and dyskinesia. Gardella and colleagues (2015) recently described three unrelated families with benign infantile seizures and dyskinesia in 16 affected family members, all of whom carried an identical heterozygous SCN8A missense mutation (p.E1483K). The affected individuals presented with benign familial infantile seizures (BFIS) during the first years of life. Interictal EEG, cognition, and development of motor skills were normal in the majority of the cases. Thirty percent of affected family members had single unprovoked seizures later in life. One third of the patients developed paroxysmal dyskinetic/dystonic movements in puberty, triggered by stretching, motor initiation or by emotional stimuli. This variant did not seem be due to a founder effect, suggesting that this may be a mutation hotspot.
Other SCN8A updates. Visit our SCN8A gene page to learn more about other updates on what’s new with SCN8A and take a look at the additional topics that we have added with our revised gene page content, including sections on the clinical perspective, animal models, references, and research.