
Issue 17/2015. This week, we discuss three recent publications on a rare cause of severe infantile epileptic encephalopathies, two novel candidate genes for focal epilepsies, and a novel genetic cause for one of the most common brain malformations in humans.
Epilepsy Awareness. Let me stop for a second before writing about the gene discoveries of the week. November is Epilepsy Awareness Month. As a researcher and clinician in the field, I want to use this opportunity to thank everybody who is involved in bringing epilepsy out of the shadows. We know that for the epilepsies, this is always going to be an uphill battle. The epilepsies are neurological diseases that present in many different ways that can sometimes be difficult to interpret. In 1960, William G. Lennox stated that epilepsy is no longer the Cinderella of Medicine, looking back at centuries of confusion with regards to the nature and cause of epilepsies. In 2015, we have many promising strategies that will hopefully help us understand and treat epilepsy better. Finding genes for epilepsies and understanding the mechanisms behind seizure disorders is our contribution to this. Making people aware of epilepsy is a major aspect of making sure that we have sufficient momentum to go forward. Here are our updates of the week.
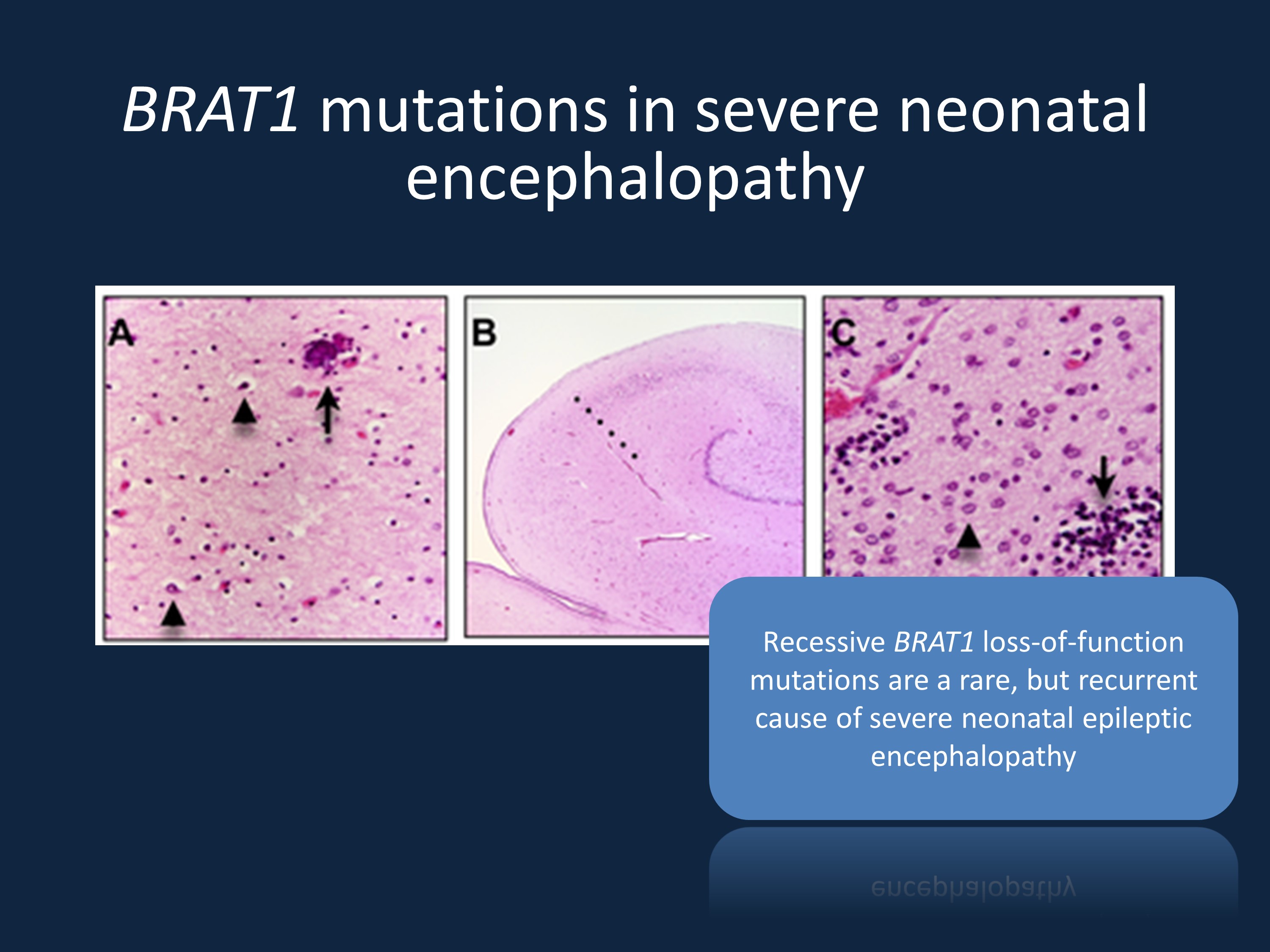
Figure and figure legend modified from Puffenberger and collaborators under a Creative Commons license (http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0028936). The sections show the histological changes from an infant who died from a homozygous BRAT1 mutation. The histological findings are compatible with corticobasal degeneration in the brain (A) In the frontal, occipital and temporal cortex, there is marked neuronal loss, gliosis with astrocytes (arrowheads) and swollen oligodendroglia. The arrow indicates a perivascular microcalcification (superior frontal gyrus, deep cortex, 10×). (B) The anterior hippocampus is smaller than expected and there is neuronal loss and gliosis in zone CA-1 (Sommer’s sector), demarcated from the CA-2 sector by the dotted line (4×). (C) At 60× magnification, the putamen shows a paucity of neurons, abundant Alzheimer Type 2 astrocytes (arrowhead) and scattered microglial nodules (arrow).
BRAT1. In a recent study in Neuropediatrics, van de Pol and collaborators describe a family with three siblings with a severe, potentially lethal epileptic encephalopathy starting in early infancy. Exome sequencing identified a frameshift mutation in BRAT1, a gene coding for a regulator of cellular responses to DNA damage. BRAT1 mutations are a known cause of a condition referred to as rigidity and multifocal seizure syndrome. It is interesting to note that BRAT1 is a gene that slowly emerged on the scene. It was initially described by Puffenberger and collaborators alongside TUBGCP6, SNIP1, CRADD, and HARS as candidate genes for severe neurological disorders in Old Order Amish and Mennonite children. Since then, several additional families with BRAT1 mutations have been published.
Comments IH: BRAT1 mutations are a recurrent cause of severe and often lethal early infantile epileptic encephalopathies. With the increased use of genetic testing in patients with severe neonatal epilepsies, more patients are described and we are able to better delineate the full phenotypic spectrum, including patients with a less severe presentation.
NPRL2, NPRL3. The mTOR cascade emerges as a key pathway for focal epilepsies, with DEPDC5 being the most prominent gene in this group of epilepsies. In a recent study in Annals of Neurology, Ricos and collaborators examine the role of the mTOR regulators NPRL2 and NPRL3 in human focal epilepsies. The authors identify mutations in both genes in roughly 1% of patients with focal epilepsies and both genes emerge as the main candidates in two families with multiple affected individuals.
Comments IH: While the evidence for both genes is not as strong as for DEPDC5, it will be interesting to see whether additional, independent studies validate the role of these genes in focal epilepsies. Both genes are ideal candidates based on their role in the mTOR pathway.
PIK3R2. Identification of genes for brain malformations is a promising branch of neurogenetics. In a recent study in Lancet Neurology, Mirzaa and collaborators identify mutations in PIK3R2 in 19 patients with bilateral perisylvian polymicrogyria (BPP), the most common form of polymicrogyria. PIK3R2 is a regulatory subunit of the PI3K-AKT-mTOR pathway. Genes for this brain malformation were previously largely elusive. The fact that many patients showed mosaicism for the mutations adds another interesting twist to this story and may make these conditions challenging for genetic counseling.
Comments IH: PIK3R2 is the most common genetic cause of BPP known to date. Mosaic mutations continue to pose a major challenge for exome sequencing. The publication by Mirzaa and collaborators emphasizes that mosaicism may be a common mechanism in some conditions and a standard exome analysis would likely miss these mutations.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.
