
Comments. After posting our 2015 update on what you should know about SCN1A, we received a number of comments on our blog and by email. We usually have the policy to respond to every comment individually. However, after we had realized that we had fallen behind with a few replies for several weeks, we felt that it might be worthwhile rephrasing some of the questions as general topics to write about, especially since many of your questions raised interesting points. Here are the questions that you asked regarding SCN1A and Dravet Syndrome.
Format. The readership of our blog is a mixture of health care professionals, scientists, patients and patient families, and lay readers. What I have tried to do in this blog post was to arrange the questions that you asked by topic, rephrase them in a more general way, and answer them as best as I could. I’d be happy for feedback regarding this new “format”. Please get back to us if you feel that your initially question had not been answered sufficiently.
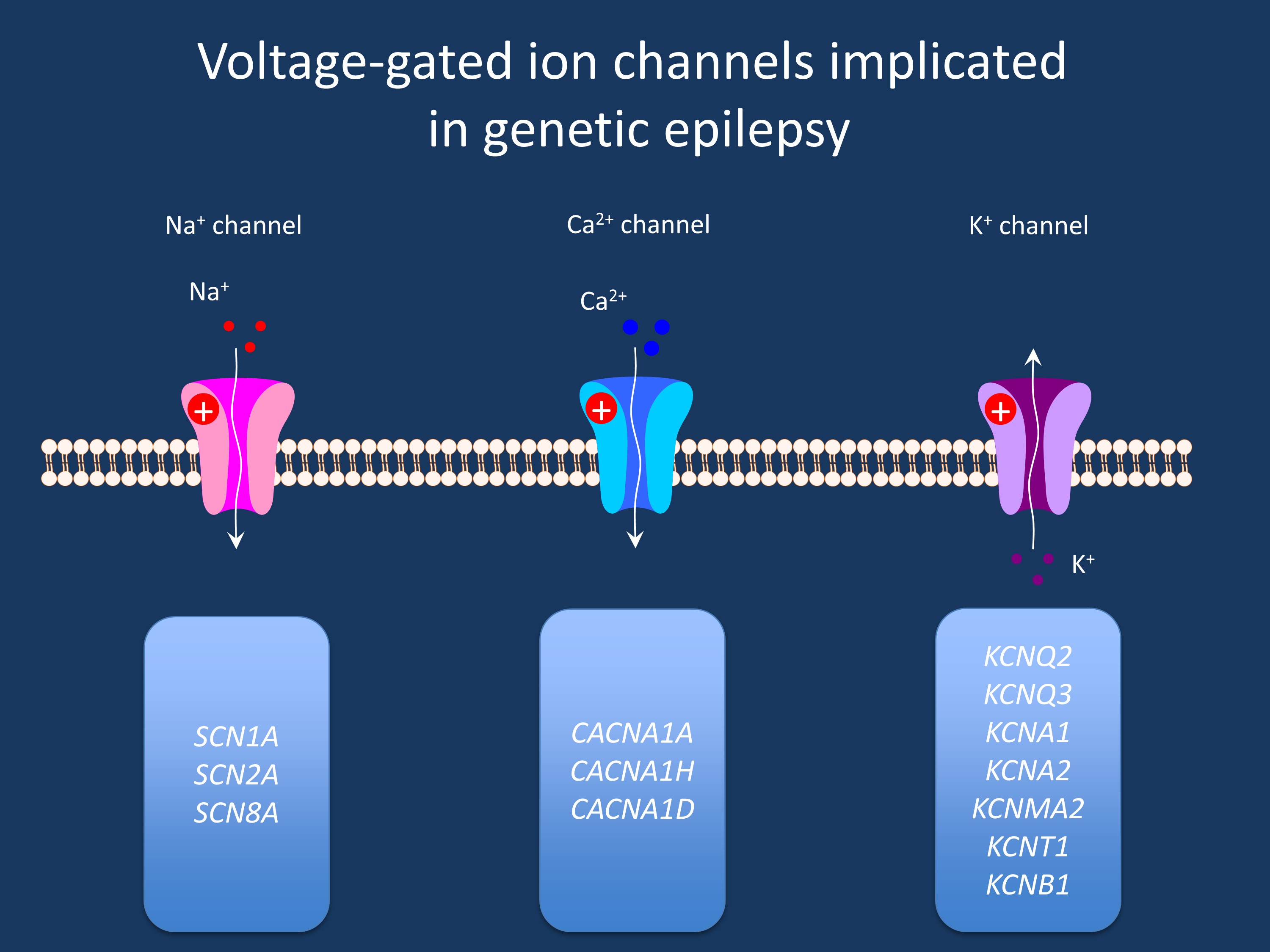
Question: How is it possible that there are unaffected individuals with SCN1A truncating mutations?
Answer: There are some families with obviously pathogenic SCN1A mutations such as truncation mutations where the mutation is inherited through unaffected parents. Given that deleterious mutations in SCN1A are usually associated with Dravet Syndrome and almost always arise de novo, this is somehow incomprehensible. Here are two possible answers. First, the SCN1A mutation may be present in mosaic state in the parent, i.e. only a subset of cells carry this mutation. This phenomenon has been described and is one reason why there is some (albeit very small) recurrence risk for Dravet Syndrome due to SCN1A mutations. Secondly, it may in fact be possible that deleterious SCN1A mutations are tolerated in healthy individuals due to modifying factors. In familial cases, SCN1A mutations are known for their broad phenotypic spectrum that includes unaffected carriers.
Question: Do you find SCN1A mutations in Temporal Lobe Epilepsy or West Syndrome?
Answer: Would we be surprised to learn about patients with mutations in SCN1A and other phenotypes, such as West Syndrome or Temporal Lobe Epilepsy? Both yes and no. Yes, because I am still holding the strong belief that there is a common theme for SCN1A and that patients share some very obvious features even though they are classified into different epilepsy syndromes. No, because SCN1A is the most common epilepsy gene and atypical presentations of a common genetic cause may be more common than other genetic causes. The Epi4K study, where SCN1A came out as the single most frequent gene, is a good example for this. Nevertheless, I think it is both very important and interesting to have a closer look at these atypical phenotypes, as they may hold the key to some of the mysteries surrounding SCN1A that we don’t yet understand.
Question: Are there treatment recommendations when variants in SCN2A or SCN8A are identified?
Answer: Several readers approached us to ask about the current state of treatment recommendations based on specific genetic findings. For genes other than SCN1A, we currently have little experience with respect to treatment recommendations that would take into account the genetic variant. Here is my big hope: collecting all the data that is out there will already help us quite a bit. We are currently raising the awareness for genetic epilepsies, and more and more patients with specific genetic alterations are being identified. We will then be able to understand the disease pattern in groups of patients with homogeneous causes of their epilepsy. For the first time, we will be able to understand features that are variable and constant, and we can collect the data that we need to finally go ahead with true personalized medicine for severe epilepsies.
Question: Is Dravet Syndrome a “true” epileptic encephalopathy?
Answer: We usually refer to Dravet Syndrome and other genetic epilepsies with intractable seizures and developmental delay as epileptic encephalopathies. As it was pointed out by some of our readers, this is technically not the way that the term epileptic encephalopathy was initially used. Conceptually, “epileptic encephalopathy” refers to a disturbance of brain function due to epileptic activity that is reversible once the epileptic activity is controlled. For example, Landau-Kleffner-Syndrome or Electrical Status Epilepticus in Slow-Wave Sleep (ESES) are examples for this. For most severe genetic epilepsies, however, the story is probably more complicated, as they are complex neurodevelopmental disorders where the neurodevelopmental issues are due to the underlying genetic defect, but not necessarily entirely reversible with good seizure control. Often, however, the complex interplay between seizure burden, seizure control, and development is not well understood. This is the reason why we refer to these conditions as epileptic encephalopathies in daily practice.
Question: Are there plans to write similar blog posts for other genes?
Answer: Yep, we’re on it. It just takes a bit of time, but I hope that we can roll out a “this is what you need to know” format for other genes including PCDH19, CHD2, DNM1 and others.
Ingo Helbig is a child neurologist and epilepsy genetics researcher working at the Children’s Hospital of Philadelphia (CHOP), USA.

Thanks a lot, Dr. Helbig!
Ulrich Stephani