
Parallel worlds. There are two fields of genetics for neurodevelopmental disorders that currently produce large amounts of data – the field of copy number variation analysis and the field of exome sequencing. When assigning pathogenicity, information from both genetic technologies are rarely considered jointly. A recent study in Nature Genetics now performs a combined analysis of a large CNV and exome datasets in intellectual disability and autism. Interestingly, this method produces robust results, highlighting novel causative genes.
Large scale data. Before delving into the discussion of the data, there is one observation to be made. The recent publication by Coe and collaborators is impressive when it comes to numbers – a good reminder that we are slowly reaching a new level in genetics where massive data alone has the chance to guide us. The authors refer to this as a genotype-first analysis, but it is actually more than this. Particularly with sample sizes in the 30,000 – 50,000 patients genotyped for CNVs, research is arriving at a stage where novel types of analyses become feasible. For example, while much is known about recurrent microdeletion hotspots, systematically findings overlaps in non-recurrent microdeletions has always been difficult. As the authors point out, the current sample sizes now allow for such an assessment. Likewise, robust analyses for modifier CNVs only become possible with such sample sizes, as demonstrated by the 16p12.1 microdeletion. In a nutshell, we’re entering the age of big data in genetics.
Combined analysis. By mining CNV data from almost 30,000 patients with autism and intellectual disability and 20,000 unaffected controls, the authors are able to highlight a number of regions that are significantly associated with neurodevelopmental disorders. These regions include the known recurrent microdeletion hotspots, for example 15q13.3 and 16p13.11. In addition, for the first time ever, a genome-wide analysis is possible to identify non-recurrent microdeletions. Overall, 14 regions make the cut. However, in many regions, the smallest possible overlap still included several genes. This is when the authors turn to available exome data.
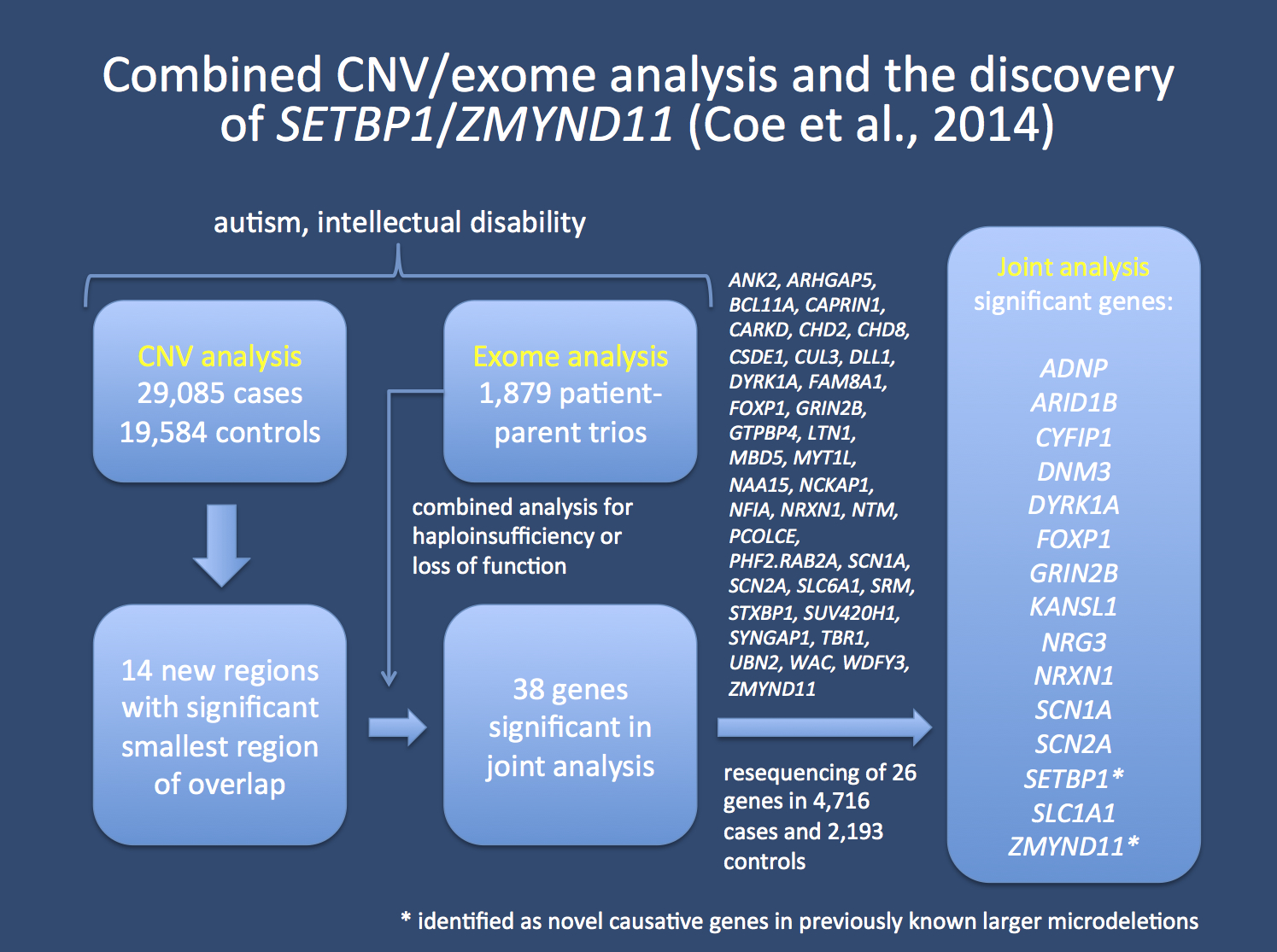
Study by Coe and collaborators combining information from CNV and exome studies. 38 genes were significant in the joint analysis, and resequencing 26 of these genes in 4,700 patients and 2,200 controls identified 15 genes that were significant. Interestingly, for two genes, namely SETBP1 and ZMYND1, the resequencing contributed to pinpointing the causative gene in a previously larger contiguous gene syndrome.
Exomes. The exome data of almost 1,300 patient-parent trios are available for analysis. The authors added the information for de novo mutations and performed an interesting joint analysis of CNV and exome data. Basically, the idea behind this combined analysis is the assumption that microdeletions may hint at genes that cause neurodevelopmental disorders when haploinsufficient, i.e. when one copy of this gene is absent. A similar effect would be predicted if there are deleterious mutations in such a gene and these mutations can be identified by exome data. By focussing on de novo mutations, the authors avoid much of the flood of genetic variants that we need to browse through when parental exomes are not available. Compared to the large CNV datasets, the statistical power of the exome data is limited. However, in the study by Coe and collaborators it was sufficient to highlight 38 genes that were significant in the joint analysis. 26 of these genes were taken to the next step, a resequencing of these genes in almost 5,000 additional cases and more than 2,000 controls.
Resequencing. The final step of the validation eventually made it possible for 15 genes to rise above the genomic noise. Many of these genes are well-known to cause intellectual disability or autism, such as DYRK1A, KANSL1 or NRXN1. Other genes were more suprising such as FOXP1, a gene previously described in intellectual disability, but not considered highly relevant. Interestingly, it produces a phenotype with a predominant speech apraxia phenotype as seen in FOXP2. In addition, SLC1A1 was significant, a previously described gene for neurodevelopmental disorders. EAAT3, the transporter encoded by SLC1A1 is a prominet glutamate transporter in neurons. DNM3 coding for dynamin 3 is related to DNM1, which was recently discovered as the underlying cause of 2% of patients with Infantile Spasms or Lennox-Gastaut Syndrome. In addition, for two genes, the resequencing effort help pinpointing the causative genes in larger contiguous gene syndromes.
SETBP1, ZMYND11. The authors identified de novo mutations in patients and compared the phenotypic overlap to patients with microdeletions spanning these genes. ZMYND11 is a candidate gene for the 10p15.3 deletion syndrome and patients showed an interesting overlap of autism, aggression and complex neuropsychiatric features. SETBP1 loss-of-function mutations were found in patients with intellectual disability and language problems. Interestingly, gain-of-function de novo mutations in this gene were previously identified in Gideon-Schinzel-Syndrome, a genetic intellectual disability syndrome that has characteristic, but entirely different dysmorphic features compared to patients with deletions or loss-of-function mutations.
This is what you need to know. By using large genetic datasets, Coe and collaborators were able to implicate several new syndromes in the genetics of autism or intellectual disability. Their combined analysis provides an interesting template for future studies, also for epileptic encephalopathies. Of note, the analysis performed by Coe and collaborators focussed on statistical significance rather than absence of mutations in controls. Therefore, many of these genes were implicated even though a deletion or mutation was found in an unaffected control – again a shift of paradigms, replacing a black-and-white concept of monogenic disease by a tour-de-force through sheer numbers.
Child Neurology Fellow and epilepsy genetics researcher at the Children’s Hospital of Philadelphia (CHOP), USA and Department of Neuropediatrics, Kiel, Germany

