
Torsional stress. The DNA double helix has one major problem that we know from telephone cords: it is difficult to untangle. However, our DNA is constantly twisted and untangled for gene transcription. This constant twisting and untwisting produces torsional stress that is relieved by topoisomerases. A recent publication in Nature Genetics now identified a human neurological phenotype that is caused by faulty activity of this mechanism: neurodegeneration with epileptic encephalopathy. However, there are some features of the phenotype that are not easily explained by erroneous DNA twisting.
The topoisomerase cascade. Torsional stress of the DNA may result in DNA breaks and problems with transcription. The DNA maintenance machinery of the cell prevents this by releasing tensional stress through topoisomerases, enzymes that cut DNA, release the torsional stress and repair the strand. It’s like cutting the tangled telephone cable at each twist and gluing the cord together again. In order for topoisomerases to work, they need to break the existing double-strand temporarily. For the topoisomerase 2, which allows for double-strand break, this is a potentially dangerous step, as the process can get interrupted. In this case, the enzyme gets stuck to the broken DNA. This is when the tyrosyl DNA phosphodiesterase-2 enzyme encoded by TDP2 comes into play; this enzyme rescues the interrupted topoisomerase enzyme function and allows for DNA repair. Gomez-Herreros and colleagues identify homozygous mutations in a family with neurodegeneration with epilepsy, suggesting that the Central Nervous System has a particularly high demand for these rescue operations.
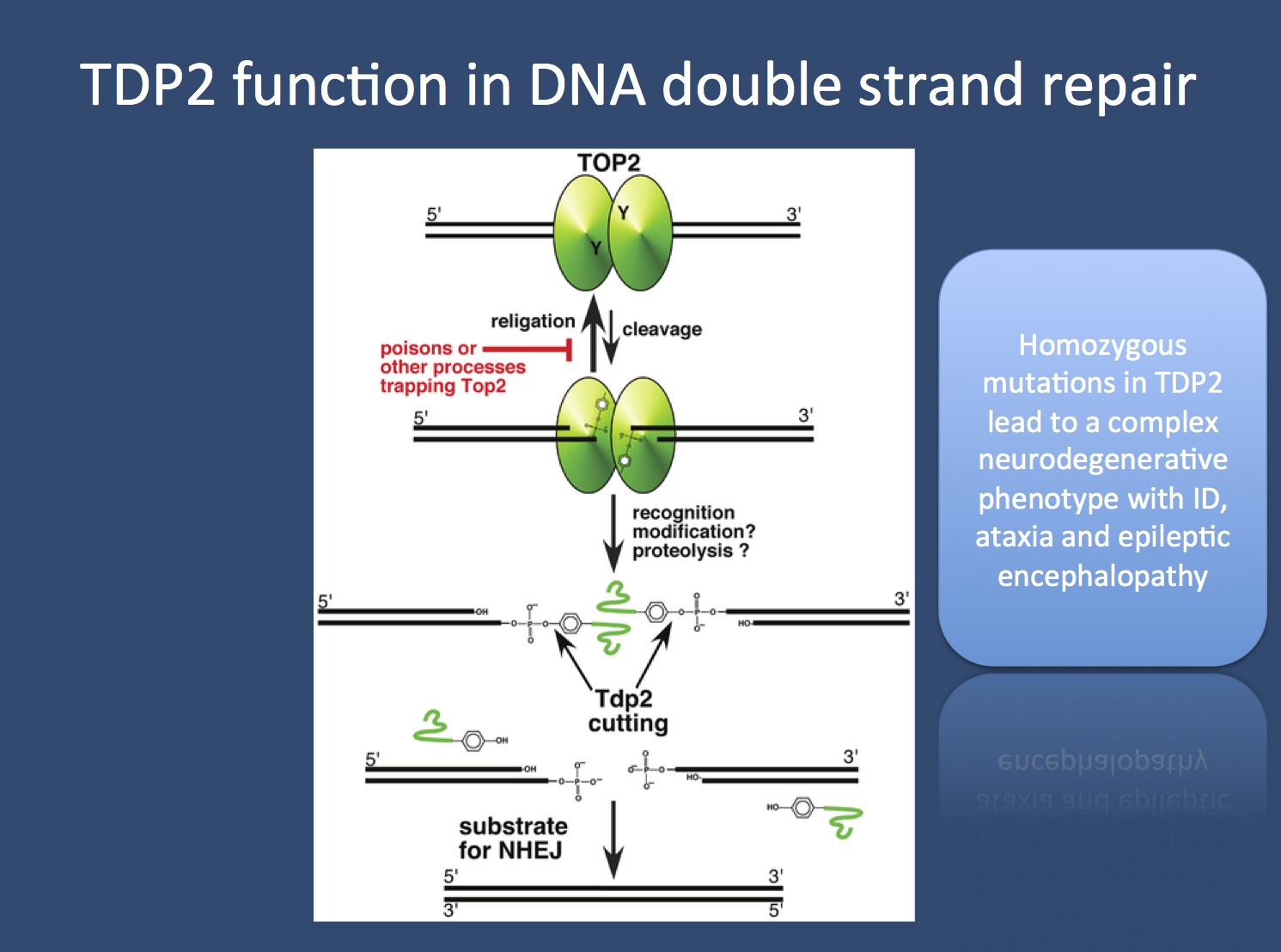
TDP2 function in DNA double strand repair. The topoisomerase 2 enzyme is important for relieving torsional stress of the DNA double helix by introducing brief double strand breaks. In some cases, the topoisomerase enzyme gets stuck to the DNA and needs to be rescued, which is performed by the tyrosyl DNA phosphodiesterase-2 enzyme encoded by the TDP2 gene. Mutations in TDP2 result in neurodegeneration with epilepsy. (Figure adapted from Nitiss et al. under a Creative Commons License http://www.plosgenetics.org/article/info:doi/10.1371/journal.pgen.1003370)
The TDP2 phenotype.Gomez-Herreros and colleagues identified homozygous mutations in TDP2 by searching for the causative gene in a consanguineous family with three affected individuals. The authors performed homozygosity mapping and exome sequencing, which resulted in the identification of a homozygous splice site mutation in TDP2. The authors went on to show that this splice mutation leads to a complete loss of available TDP2 protein, most probably by introducing a premature stop codon. All three affected family members had some minor dysmorphic features and a distinct neurological phenotype. All three patients had essentially normal neurological development until the age of 2 with subsequent neurodegeneration. In adulthood, all patients had ataxia, were largely wheelchair-bound and had refractory epilepsy. Seizures usually started in infancy with a notable seizure-free period in childhood and subsequent epileptic encephalopathy.
Gene transcription. Gomez-Herreros and colleagues continued to assess the function of TDP2 in mice. They found that the gene is expressed in brain, but TDP2-deficient animals did not have any observable phenotypes. However, upon histological examination, the number of cerebellar interneurons was markedly reduced. This suggests that these types of cells are particularly vulnerable to TDP2 deficiency. Also, the vulnerability of interneurons may explain the epileptic encephalopathy in patients with TDP2 mutations. When the authors examined gene expression profiles in the brains of TDP2 deficient animals, they identified more than 160 genes that were dysregulated. Most genes were downregulated, which suggests that lack of TDP2 largely leads to a reduced expression of crucial genes. Amongst these genes were several genes for GABA receptors including GABBR2, GABRA5, GABRG1 and GABRG3, a group of genes that somewhat stood out in the list of dysregulated transcripts.
Summary. Erroneous DNA repair may result in primary neurological dysfunction without largely affecting other organ systems. This finding is surprising as most DNA repair defects such as Xeroderma pigmentosum mainly affect other organs with a frequent cell turnover without a prominent effect on primary neurological functioning. However, a subset of DNA repair mechanisms and defects of the DNA maintenance machinery seem to primarily affect function of the nervous system. For example, this is known for mutations affecting the PNKP gene, which also result in a progressive neurodegenerative disease and epileptic encephalopathy. A similar picture seems to emerge for the human phenotypes due to TDP2 mutations.
Child Neurology Fellow and epilepsy genetics researcher at the Children’s Hospital of Philadelphia (CHOP), USA and Department of Neuropediatrics, Kiel, Germany

