
That Sam I Am. The entire field of high-throughput genomics appears to be inspired by the American children’s book author Dr. Seuss. Given that we are currently reading through the original books almost on a daily basis due to the presence of a toddler in our home, mentioning *.sam files, *.bam files or sam2bam routines always makes me smile. However, this is not a post about children’s books; it’s about a likely 2013 trend in genomic research, the redefinition of the boundary between genome center and end user and the laptopification of life sciences.
The 2013 challenge. Looking at 2013 unfolding, there will probably two trends. First, exome sequencing will slowly replaced by genome sequencing in cutting edge genomic science. Secondly, as exome sequencing has become a standard procedure, the analysis and interpretation of exome data will shift from genome centers to individual scientists as has happened before with the analysis of copy number variations. During the holidays, I tried to explore the possibility to analyze exomes on my laptop. This is the first post describing my experience with the three essential tools for exome analysis in patient-parent trios: samtools, DeNovoGear and annovar. Samtools is a bioinformatic workhorse to cut, format and extract data from large BAM files, DeNovoGear is the main program for the detection of de novo mutations, and annovar is the standard annotation tool, i.e. the program that translates chromosomal coordinates into positions within genes including the impact of a given mutation. Let’s start with samtools.
BAM, here’s your exome. The BAM file will probably be the ideal intersection between genome center and exome data end user. BAM files are large binary files that contain the aligned sequenced reads. Massive parallel sequencing produces a large number of small sequencing reads that have to be mapped to the reference human genome. This process, read mapping, is computationally intensive and –in my impression- well beyond the reach of a non-bioinformatician. However, the read mapping is stored in a specific file format called Sequence Alignment/Map format or SAM format. Its binary, i.e. compressed and harddrive-space saving, counterpart is the BAM file. A BAM file is usually around 5-8 GB for a single exome, the size of a medium-sized iTunes library. If you get the original BAM files from your genome center in addition to the variant files, you can already do quite a bit.
Samtools. For the analysis of exome data, BAM files need to manipulated or extracted in various ways. The standard program for this is samtools. Samtools is a program running under a Unix environment, i.e. you can either run it on a Linux computer, use the Mac OS Terminal or Cygwin on Windows computers. In addition to the BAM files, samtools needs the original genome files that the BAM files were aligned to. This might seem paradoxical in the first place, but there are many different versions of the human genome floating around in present day genome centers and for samtools, we need the original human genome file that was used for the mapping. This means that you need another 3.5 GB of data (the human genome FASTA file) in addition to the BAM files. But beware, if you try to use the wrong FASTA file, samtools cannot operate. This is a lesson I had to learn the hard way. So what can you do with samtools?
Coverage in a specific genomic region. For a recent project, we tried to see how good a certain genomic region is covered with existing exome data. For example, if we want to exclude SLC2A1 mutations, we can either sequence the gene or –if available- look at the read depth of exome data. In previous posts last year, we emphasized the fact that exome data has gaps and cannot be used to exclude mutations (“The Exome Fallacy”). However, this is a very global statement and some genes might be covered quite well. To find out how good the coverage is, we can use samtools to get the read depth at each base pair of the coding region. You can extract the genomic coordinates of the exons through the CCDS Database and then transform the BAM file into a variant call file or VCF file. VCF files unfortunately have the same ending as the files used by address book programs, so don’t doubleclick on it. The command used by samtools is the infamous mpileup, a command to manipulate large Gigabyte files that even starts piling up data if the input format is wrong (i.e. you waste 6-8 hours to find out that it didn’t work). Using mpileup and bcftools for exon 1 of SLC2A1, we get a VCF file for each base pair of exon 1. The VCF file can then be imported into Excel and the read depth can be extracted. I then used a small R script to plot the coverage in the exon and the surrounding 5’UTR and intron sequence (see below).
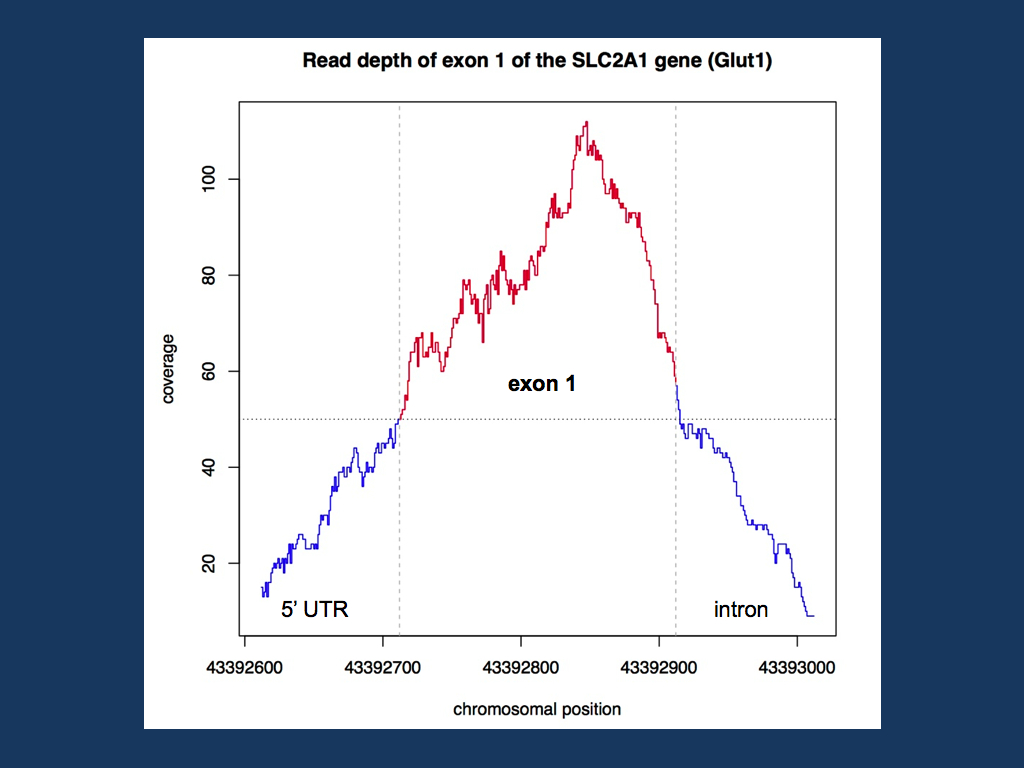
Coverage of the first exon of the SLC2A1 gene (Glut1) in a random EuroEPINOMICS exome. The first exome of this gene is well covered with a read depth of 50 and more at each base pair of the exon.
Full coverage. Exon 1 of the SLC2A1 gene is well covered in the exome that I have analyzed here. The entire exonic sequence has a read depth of at least 50, which should be sufficient to identify deviations from the reference sequence, i.e. possible mutations. In addition to the actual coverage, the mapping quality should be taken into account (I am still working on this…). But in our example, samtools was the pivotal tool to extract the full sequence for a region of interest from a BAM file. This is the technique that can be used to query exome data for the coverage of known epilepsy genes.
Child Neurology Fellow and epilepsy genetics researcher at the Children’s Hospital of Philadelphia (CHOP), USA and Department of Neuropediatrics, Kiel, Germany

